

Molecular and Pathological Analyses of IARS1-Deficient Mice: An IARS Disorder Model

 and
and
Abstract
:1. Introduction
2. Results
2.1. Introduction of the V79L Mutation Using CRISPR/Cas9
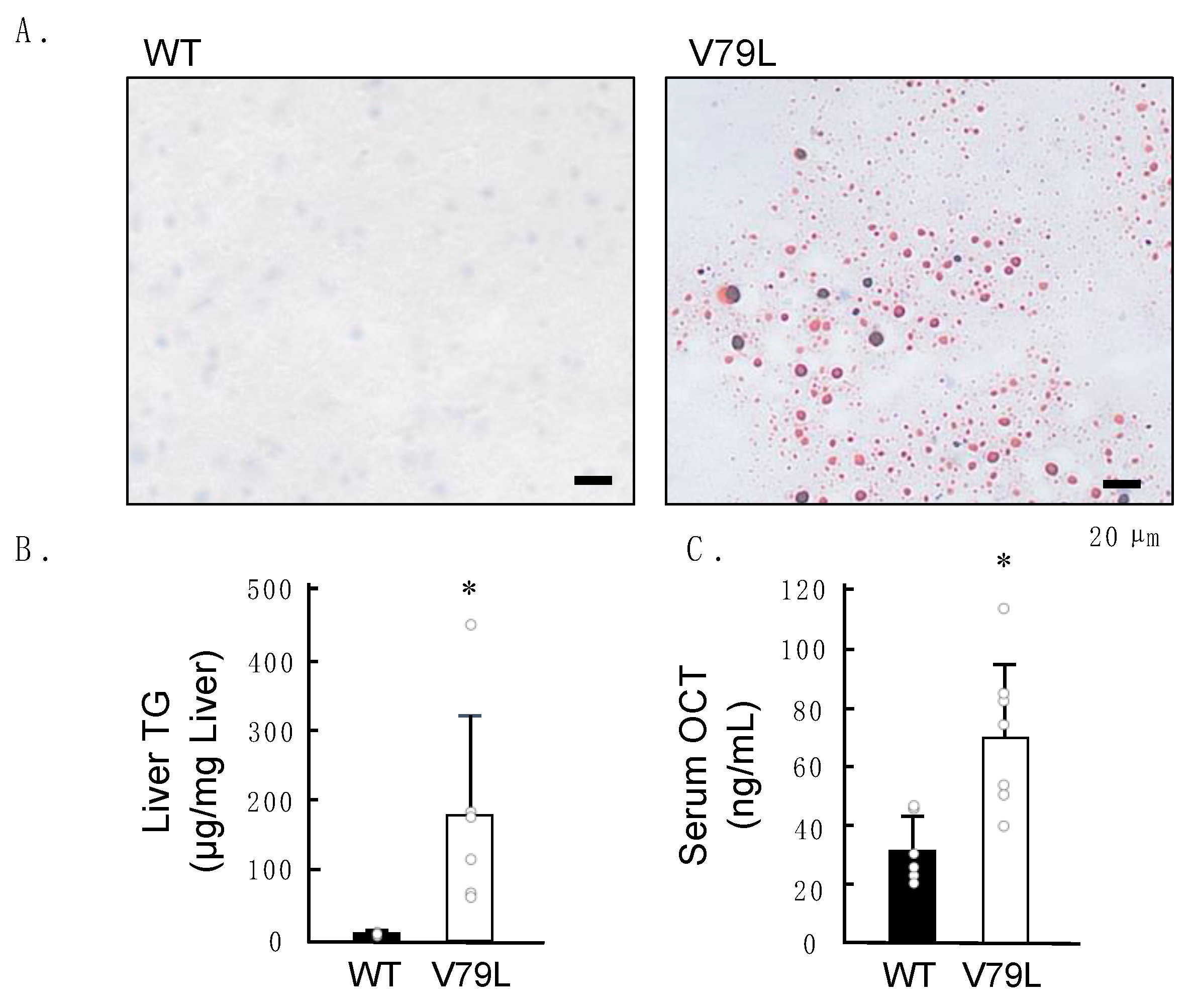
2.2. Histopathological Observation of Mice Liver
2.3. Elevation of Hepatic Triglycerides and Serum Ornithine Carbamoyltransferase Levels in Mutant Mice
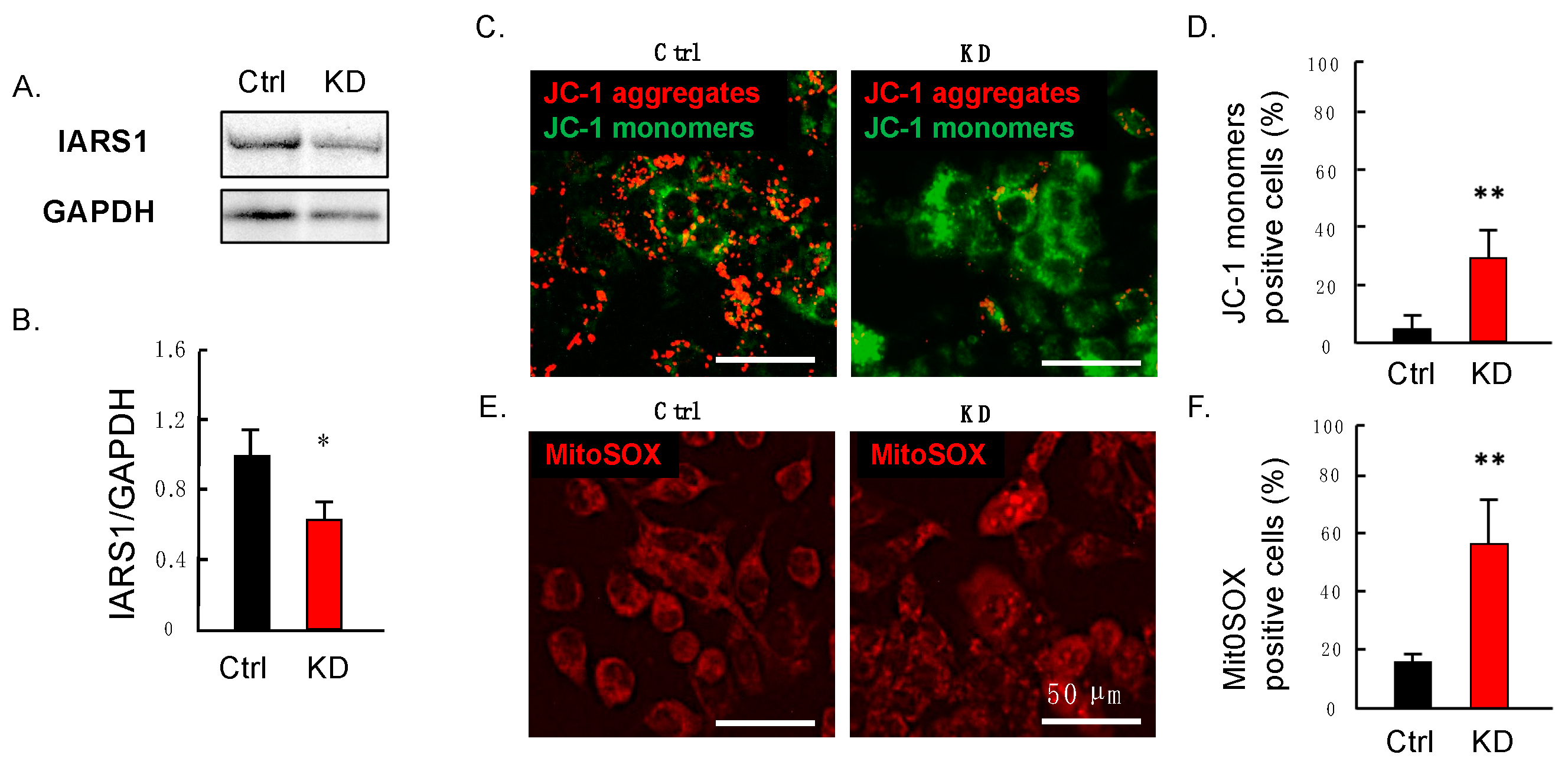
2.4. Downregulation of IARS1 Induces Mitochondrial Dysfunction in HepG2 Cells
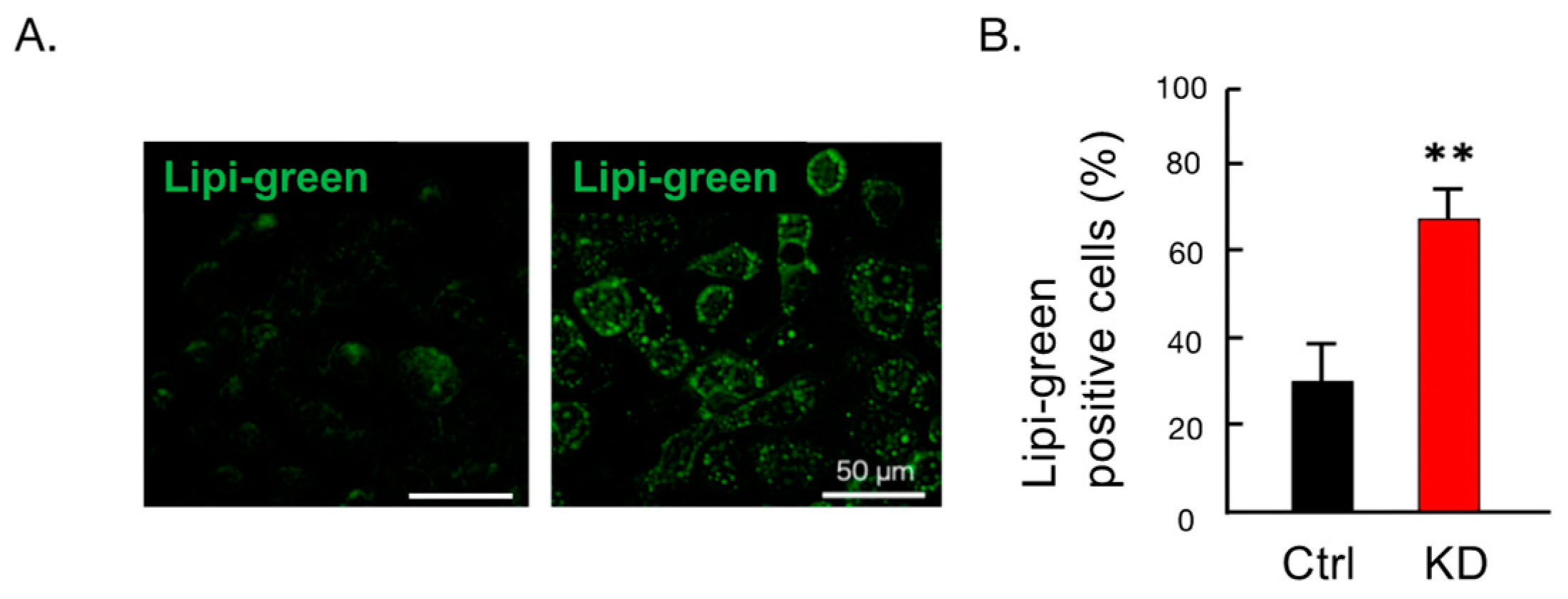
2.5. Administration of Palmitic Acid Increased Fat Droplet Formation in HepG2 Cells
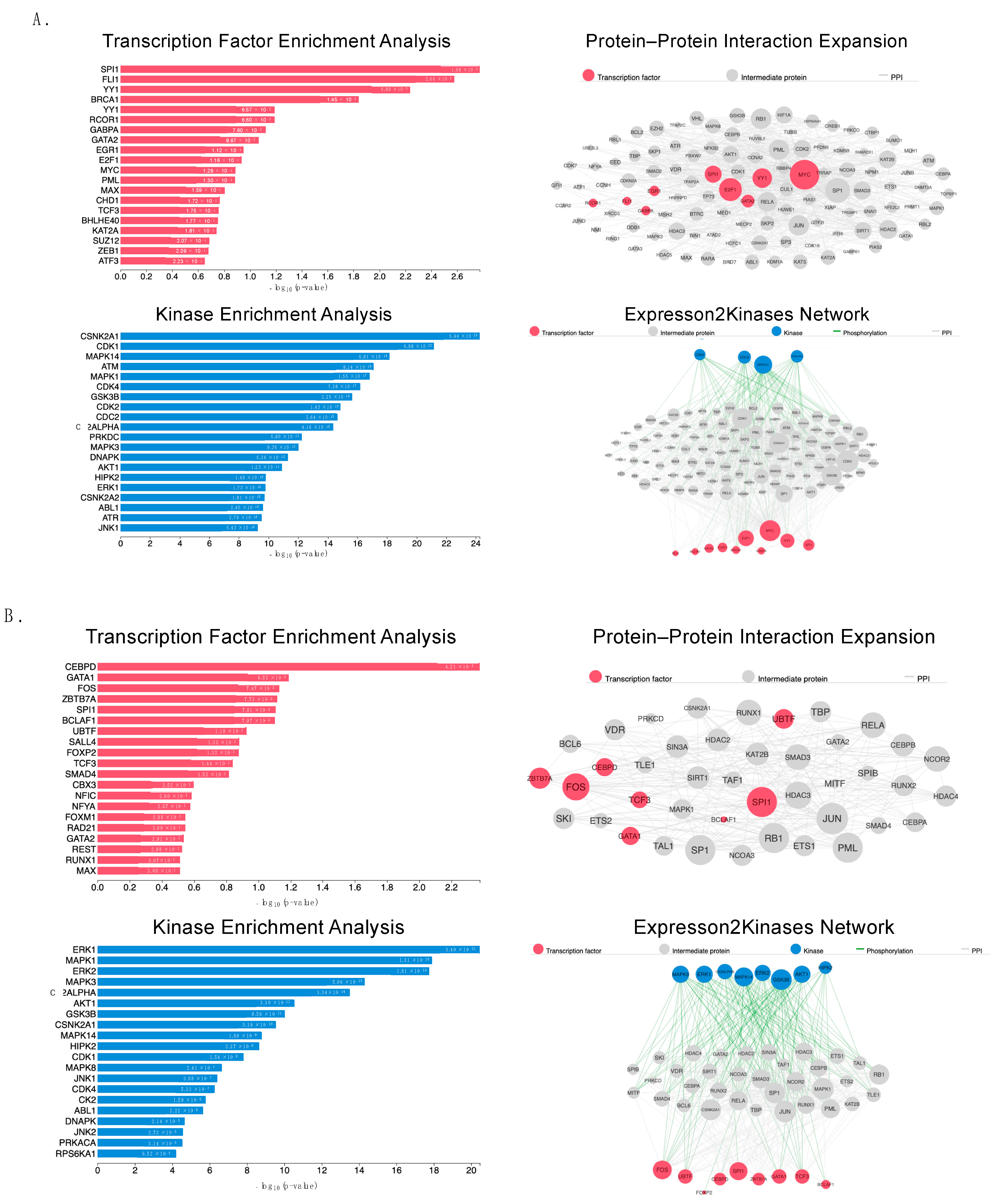
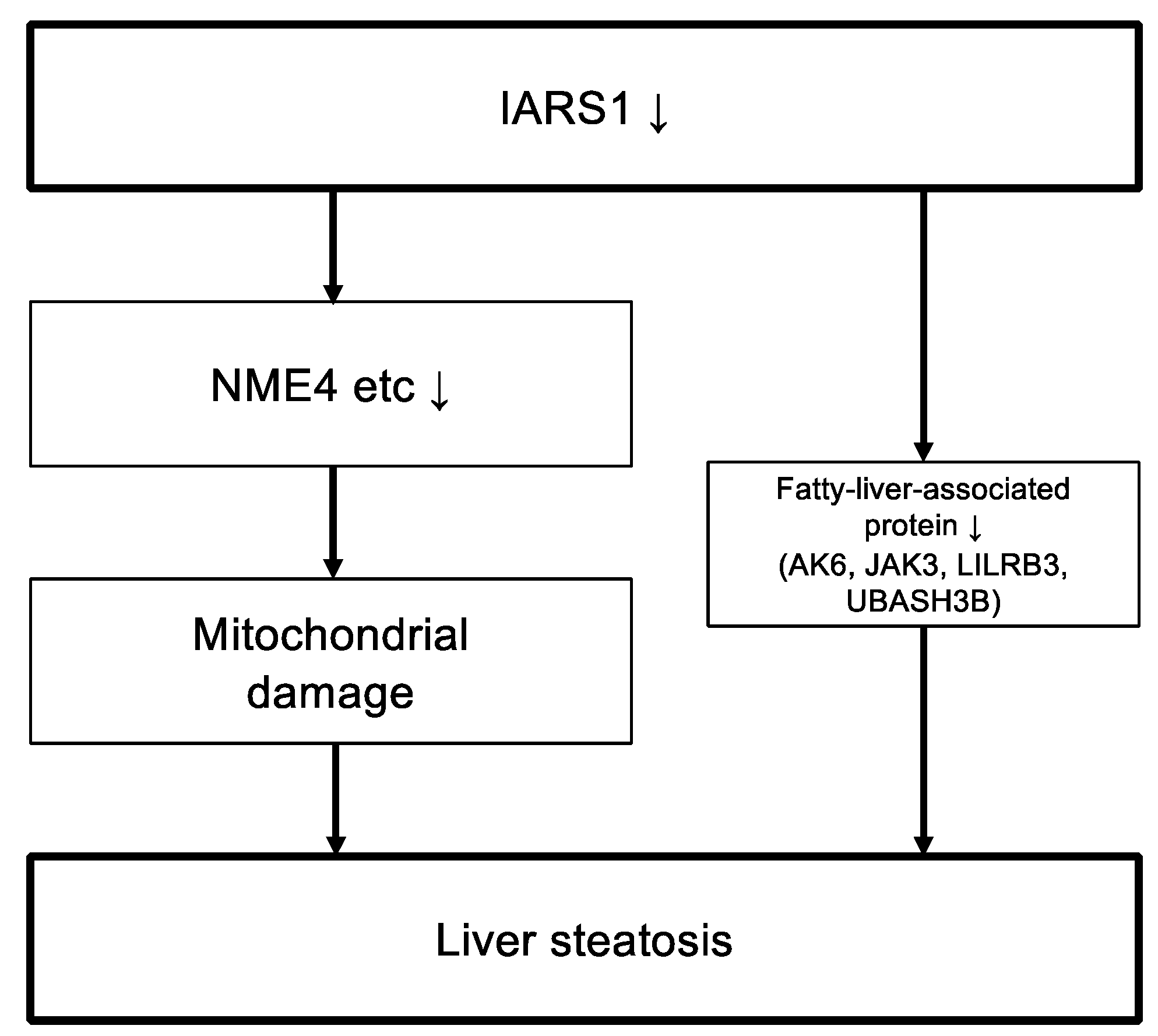
2.6. Proteomic Analysis
3. Discussion
4. Materials and Methods
4.1. Ethical Statements
4.2. Animals
4.3. Western Blotting
4.4. Histology
4.5. Estimation of Liver TG Level
4.6. Measurement of Serum OCT Levels
4.7. Cell Culture and Silencing of IARS1 Gene
4.8. Estimation of the Mitochondrial Membrane Potential and ROS
4.9. Treatment with Palmitic Acid
4.10. Proteomics
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Mccormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update. Curr. Opin. Pediatr. 2018, 30, 714. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Hanaford, A.R.; Cho, Y.; Nakai, H. AAV-vector based gene therapy for mitochondrial disease: Progress and future perspectives. Orphanet. J. Rare Dis. 2022, 17, 1–21. [Google Scholar] [CrossRef]
- Ayers, M.; Horslen, S.P.; Gómez, A.M.; Squires, J.E. Mitochondrial hepatopathy. Clin. Liver Dis. 2022, 26, 421–438. [Google Scholar] [CrossRef]
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef]
- Hirano, T.; Kobayashi, N.; Matsuhashi, T.; Watanabe, D.; Watanabe, T.; Takasuga, A.; Sugimoto, M.; Sugimoto, Y. Mapping and exome sequencing identifies a mutation in the IARS gene as the cause of hereditary perinatal weak calf syndrome. PLoS ONE 2013, 8, e64036. [Google Scholar] [CrossRef] [Green Version]
- Ogata, Y.; Nakao, T.; Takahashi, K.; Abe, H.; Misawa, T.; Urushiyama, Y.; Sakai, J. Intrauterine growth retardation as a cause of perinatal mortality in Japanese black beef calves. Zentralbl. Veterinarmed. A 1999, 46, 327–334. [Google Scholar] [CrossRef]
- Kopajtich, R.; Murayama, K.; Janecke, A.; Haack, T.; Breuer, M.; Knisely, A.S.; Harting, I.; Ohashi, T.; Okazaki, Y.; Watanabe, D.; et al. Biallelic IARS mutations cause growth retardation with prenatal onset, intellectual disability, muscular hypotonia, and infantile hepatopathy. Am. J. Hum. Genet. 2016, 99, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, N.; Weiss, K.; Oprescu, S.N.; Shapira, R.; Kidron, D.; Vanagaite-Basel, L.; Antonellis, A.; Muenke, M. Bi-allelic IARS mutations in a child with intra-uterine growth retardation, neonatal cholestasis, and mild developmental delay. Clin. Genet. 2017, 91, 913. [Google Scholar] [CrossRef] [PubMed]
- Murayama, H.; Ikemoto, M.; Fukuda, Y.; Tsunekawa, S.; Nagata, A. Advantage of serum type-I arginase and ornithine carbamoyltransferase in the evaluation of acute and chronic liver damage induced by thioacetamide in rats. Clin. Chim. Acta 2007, 375, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Murayama, H.; Ikemoto, M.; Fukuda, Y.; Nagata, A. Superiority of serum type-I arginase and ornithine carbamyltransferase in the detection of toxicant-induced acute hepatic injury in rats. Clin. Chim. Acta 2008, 391, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Crudele, A.; Panera, N.; Romito, I.; Meroni, M.; De Stefanis, C.; Palma, A.; Comparcola, D.; Fracanzani, A.L.; Miele, L.; et al. Β-Klotho gene variation is associated with liver damage in children with NAFLD. J. Hepatol. 2020, 72, 411–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Dou, X.; Ning, H.; Song, Q.; Wei, W.; Zhang, X.; Shen, C.; Li, J.; Sun, C.; Song, Z. Sirtuin 3 Acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 2017, 66, 936. [Google Scholar] [CrossRef] [Green Version]
- Indo, H.P.; Davidson, M.; Yen, H.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef]
- Takashi, Y.; Tomita, K.; Kuwahara, Y.; Roudkenar, M.H.; Roushandeh, A.M.; Igarashi, K.; Nagasawa, T.; Nishitani, Y.; Sato, T. Mitochondrial dysfunction promotes aquaporin expression that controls hydrogen peroxide permeability and ferroptosis. Free Radic. Biol. Med. 2020, 161, 60–70. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [Green Version]
- Clarke, D.J.; Kuleshov, M.V.; Schilder, B.M.; Torre, D.; Duffy, M.E.; Keenan, A.B.; Lachmann, A.; Feldmann, A.S.; Gundersen, G.W.; Silverstein, M.C.; et al. eXpression2Kinases (X2K) Web: Linking expression signatures to upstream cell signaling networks. Nucleic. Acids Res. 2018, 46, W171–W179. [Google Scholar] [CrossRef] [Green Version]
- Boison, D.; Scheurer, L.; Zumsteg, V.; Rülicke, T.; Litynski, P.; Fowler, B.; Brandner, S.; Mohler, H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc. Natl. Acad. Sci. USA 2002, 99, 6985–6990. [Google Scholar] [CrossRef] [Green Version]
- Mishra, J.; Verma, R.K.; Alpini, G.; Meng, F.; Kumar, N. Role of janus kinase 3 in predisposition to obesity-associated metabolic syndrome. J. Biol. Chem. 2015, 290, 29301–29312. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Jiang, Z.; Dai, H.; Miao, R.; Shu, J.; Gu, H.; Liu, X.; Huang, Z.; Yang, G.; Chen, A.F.; et al. Hepatic leukocyte immunoglobulin-like receptor B4 (LILRB4) attenuates nonalcoholic fatty liver disease via SHP1-TRAF6 pathway. Hepatology 2018, 67, 1303. [Google Scholar] [CrossRef] [Green Version]
- Boissan, M.; Dabernat, S.; Peuchant, E.; Schlattner, U.; Lascu, I.; Lacombe, M. Mammalian Nm23/NDPK family: From metastasis control to cilia movement. Mol. Cell Biochem. 2009, 329, 51–62. [Google Scholar] [CrossRef]
- Lacombe, M.; Lamarche, F.; De Wever, O.; Padilla-Benavides, T.; Carlson, A.; Khan, I.; Huna, A.; Vacher, S.; Calmel, C.; Desbourdes, C.; et al. The mitochondrially-localized nucleoside Diphosphate kinase D (NME4) is a novel metastasis suppressor. BMC Biol. 2021, 19, 1–29. [Google Scholar] [CrossRef]
- Ma, M.; Xie, W.; Li, X. Identification of autophagy-related genes in the progression from non-alcoholic fatty liver to non-alcoholic steatohepatitis. Int. J. Gen. Med. 2021, 14, 3163–3176. [Google Scholar] [CrossRef]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA synthetase mutations in charcot-marie-tooth disease type 2d and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003, 72, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Hadchouel, A.; Wieland, T.; Griese, M.; Baruffini, E.; Lorenz-Depiereux, B.; Enaud, L.; Graf, E.; Dubus, J.C.; Halioui-Louhaichi, S.; Coulomb, A.; et al. Biallelic mutations of the methionyl-trna synthetase (MARS) cause a specific type of pulmonary alveolar proteinosis prevalent on réunion island. Am. J. Hum. Genet. 2015, 96, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.P.; McGettigan, P.; Lynam-Lennon, N.; McDermott, M.; Regan, R.; Conroy, J.; Bourke, B.; Sullivan, J.O.; Crushell, E.; Lynch, S.; et al. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol. Genet. Metab. 2012, 106, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Pang, Y.L.J.; Poruri, K.; Martinis, S.A. tRNA synthetase: tRNA Aminoacylation and Beyond. Wiley Interdiscip Rev. RNA 2014, 5, 461–480. [Google Scholar] [CrossRef] [Green Version]
- Turvey, A.K.; Horvath, G.A.; Cavalcanti, A.R.O. Aminoacyl-tRNA synthetases in human health and disease. Front. Physiol. 2022, 13, 1029218. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.V.; Lakunina, V.A.; Tarassov, I.; Krasheninnikov, I.A.; Kamenski, P.A. Noncanonical functions of aminoacyl-tRNA synthetases. Biochemistry 2012, 77, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ofir-Birin, Y.; Fang, P.; Bennett, S.; Zhang, H.; Wang, J.; Rachmin, I.; Shapiro, R.; Song, J.; Dagan, A.; Pozo, J.; et al. Structural switch of Lysyl-tRNA synthetase between translation and transcription. Mol. Cell 2013, 49, 30–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baymiller, M.; Nordick, B.; Forsyth, C.M.; Martinis, S.A. Tissue-specific alternative splicing separates the catalytic and cell signaling functions of human Leucyl-tRNA synthetase. J. Biol. Chem. 2022, 298, 101757. [Google Scholar] [CrossRef]
- Boissan, M.; Schlattner, U.; Lacombe, M. The NDPK/NME Superfamily: State of the Art. Lab. Invest. 2018, 98, 164. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, J.; Lu, Y.; Bian, H.; Yang, L.; Wu, H.; Zhang, X.; Zhang, B.; Xiong, M.; Chang, Y.; et al. DRAK2 aggravates nonalcoholic fatty liver disease progression through SRSF6-associated RNA alternative splicing. Cell Metab. 2021, 33, 2004–2020.e9. [Google Scholar] [CrossRef]
- Xu, Q.; Qi, W.; Zhang, Y.; Wang, Q.; Ding, S.; Han, X.; Zhao, Y.; Song, X.; Zhao, T.; Zhou, L.; et al. DNA methylation of JAK3/STAT5/PPARγ regulated the changes of lipid levels induced by Di (2-Ethylhexyl) phthalate and high-fat diet in adolescent rats. Environ. Sci. Pollut. Res. 2020, 27, 30232–30242. [Google Scholar] [CrossRef]
- Qin, L.; Fan, M.; Candas, D.; Jiang, G.; Papadopoulos, S.; Tian, L.; Woloschak, G.; Grdina, D.; Li, J. CDK1 enhances mitochondrial bioenergetics for radiation-induced DNA repair. Cell Rep. 2015, 13, 2056–2063. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Ying, S. CDK1: Beyond cell cycle regulation. Aging 2017, 14, 2465–2466. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- Guleria, A.; Chandna, S. ATM kinase: Much more than a DNA damage responsive protein. DNA Repair 2016, 39, 1–20. [Google Scholar] [CrossRef]
- Papeta, N.; Zheng, Z.; Schon, E.A.; Brosel, S.; Altintas, M.M.; Nasr, S.H.; Reiser, J.; D’Agati, V.D.; Gharavi, A.G. Prkdc participates in mitochondrial genome maintenance and prevents adriamycin-induced nephropathy in mice. J. Clin. Invest. 2010, 120, 4055–4064. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Takahashi, Y.; Hiura, K.; Nakano, K.; Okamura, T.; Sasaki, H.; Sasaki, N. A Single Amino Acid Substitution in PRKDC is a Determinant of Sensitivity to Adriamycin-Induced Renal Injury in Mouse. Biochem. Biophys. Res. Commun. 2021, 556, 121–126. [Google Scholar] [CrossRef]
- Smigiel, R.; Biela, M.; Biernacka, A.; Stembalska, A.; Sasiadek, M.; Kosinska, J.; Rydzanicz, M.; Ploski, R. New evidence for association of recessive IARS gene mutations with hepatopathy, hypotonia, intellectual disability and growth retardation. Clin. Genet. 2017, 92, 671. [Google Scholar] [CrossRef]
- Zou, T.; Sun, H.; Zhu, Y.; He, T.; Ling, W.; Zhu, H.; Lin, Z.; Liu, Y.; Liu, S.; Wang, H.; et al. Compound heterozygous variations in IARS1 cause recurrent liver failure and growth retardation in a Chinese patient: A case report. BMC Pediatr. 2022, 22, 1–8. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497. [Google Scholar] [CrossRef]
- Kawai, S.; Takagi, Y.; Kaneko, S.; Kurosawa, T. Effect of three types of mixed anesthetic agents alternate to ketamine in mice. Exp. Anim. 2011, 60, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Kanda, Y. Investigation of the Freely Available Easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intensity WT | Intensity V79L | Protein ID | Protein Name | Gene | Protein Description | Reference |
|---|---|---|---|---|---|---|
| Upregulated proteins | ||||||
| NaN | 18.72208977 | P70403 | CASP | Cux1 | Protein CASP | |
| NaN | 18.66755104 | Q9JMA7 | CP341 | Cyp3a41a | Cytochrome P450 3A41 | |
| NaN | 15.12169933 | Q9DBB1 | DUS6 | Dusp6 | Dual specificity protein phosphatase 6 | |
| NaN | 18.00749779 | Q920L5 | ELOV6 | Elovl6 | Elongation of very long chain fatty acids protein 6 | |
| NaN | 14.84197235 | Q9DBY0 | FOXP4 | Foxp4 | Forkhead box protein P4 | |
| NaN | 18.80181503 | A2ARV4 | LRP2 | Lrp2 | Low-density lipoprotein receptor-related protein 2 | |
| NaN | 20.06907272 | P02762 | MUP2 | Mup2 | Major urinary protein 2 | |
| NaN | 18.44208145 | Q5FW60 | MUP20 | Mup20 | Major urinary protein 20 | |
| NaN | 17.10871887 | Q80UQ2 | RASF6 | Rassf6 | Ras association domain-containing protein 6 | |
| NaN | 19.52822304 | Q9QXZ6 | SO1A1 | Slco1a1 | Solute carrier organic anion transporter family member 1A1 | |
| Downregulated proteins | ||||||
| 14.14918327 | NaN | Q8VCP8 | KAD6 | Ak6 | Adenylate kinase isoenzyme 6 | # [20] |
| 18.82424736 | NaN | Q8CJ27 | ASPM | Aspm | Abnormal spindle-like microcephaly-associated protein homolog | |
| 14.77607822 | NaN | Q60943 | I17RA | Il17ra | Interleukin-17 receptor A | |
| 14.45795155 | NaN | P23611 | IRF8 | Irf8 | Interferon regulatory factor 8 | |
| 16.59717178 | NaN | Q62137 | JAK3 | Jak3 | Tyrosine-protein kinase JAK3 | # [21] |
| 13.80301094 | NaN | P97484 | LIRB3 | Lilrb3 | Leukocyte immunoglobulin-like receptor subfamily B member 3 | # [22] |
| 12.92001438 | NaN | Q9WV84 | NDKM | Nme4 | Nucleoside diphosphate kinase, mitochondrial | $ [23,24] |
| 23.71816444 | NaN | Q8BZF8 | PGM5 | Pgm5 | Phosphoglucomutase-like protein 5 | |
| 15.65983963 | NaN | Q9CZR3 | TM40L | Tomm40l | Mitochondrial import receptor subunit TOM40B | |
| 12.56750202 | NaN | Q8BGG7 | UBS3B | Ubash3b | Ubiquitin-associated and SH3 domain-containing protein B | # [25] |
| Human | Bovine | Mouse | |
|---|---|---|---|
| Clinical features | |||
| Growth retardation Hepatopathy with steatosis Muscular hypotonia Intellectual disability Diabetes Immunodeficiency | + + + + + ND | + + + ND ND + | + (Male only) + ND ND ND ND |
| Mutation | p.[R254*]/[P437L] heterozygous [10] p.[R418*]/[I1174N] heterozygous [10] p.[V370G]/[N992D] heterozygous [10] p.[R739C]/[F556S] heterozygous [11] p.[Q671fs]/[T69I] heterozygous [44] p.[L234P]/[R519C] heterozygous [45] | p.[V79L] homozygous [8] | p.[V79L] homozygous [The present study] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, M.; Shishido, K.; Kanehira, N.; Hiura, K.; Nakano, K.; Okamura, T.; Ando, R.; Sasaki, H.; Sasaki, N. Molecular and Pathological Analyses of IARS1-Deficient Mice: An IARS Disorder Model. Int. J. Mol. Sci. 2023, 24, 6955. https://doi.org/10.3390/ijms24086955
Watanabe M, Shishido K, Kanehira N, Hiura K, Nakano K, Okamura T, Ando R, Sasaki H, Sasaki N. Molecular and Pathological Analyses of IARS1-Deficient Mice: An IARS Disorder Model. International Journal of Molecular Sciences. 2023; 24(8):6955. https://doi.org/10.3390/ijms24086955
Chicago/Turabian StyleWatanabe, Masaki, Koya Shishido, Nao Kanehira, Koki Hiura, Kenta Nakano, Tadashi Okamura, Ryo Ando, Hayato Sasaki, and Nobuya Sasaki. 2023. "Molecular and Pathological Analyses of IARS1-Deficient Mice: An IARS Disorder Model" International Journal of Molecular Sciences 24, no. 8: 6955. https://doi.org/10.3390/ijms24086955