
Interaction between Cigarette Smoke and Human Papillomavirus 16 E6/E7 Oncoproteins to Induce SOD2 Expression and DNA Damage in Head and Neck Cancer

 , and
, and
Abstract
:1. Introduction
2. Results
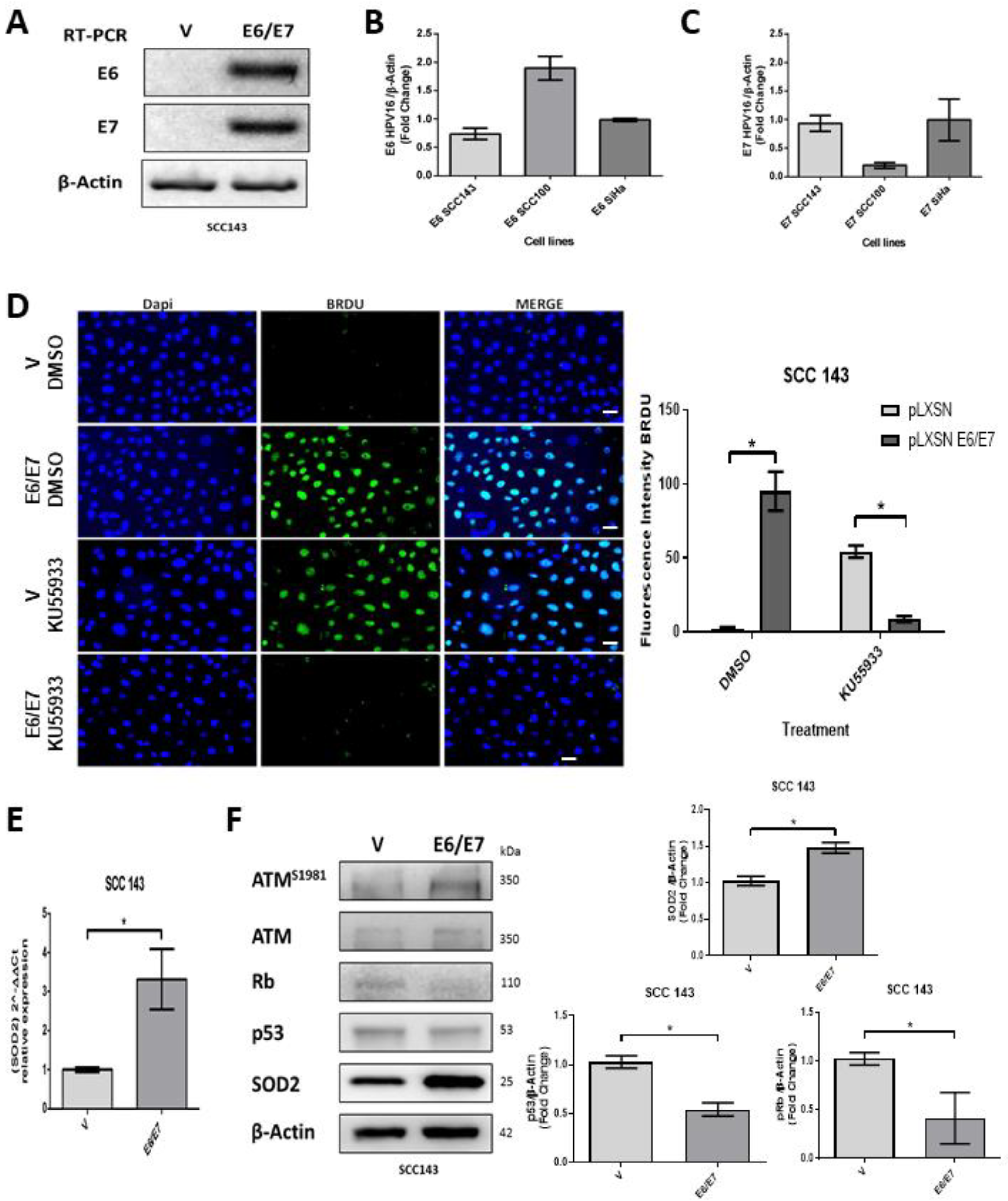
2.1. HPV16 E6 and E7 Promote SOD2 Expression in Oral Cells
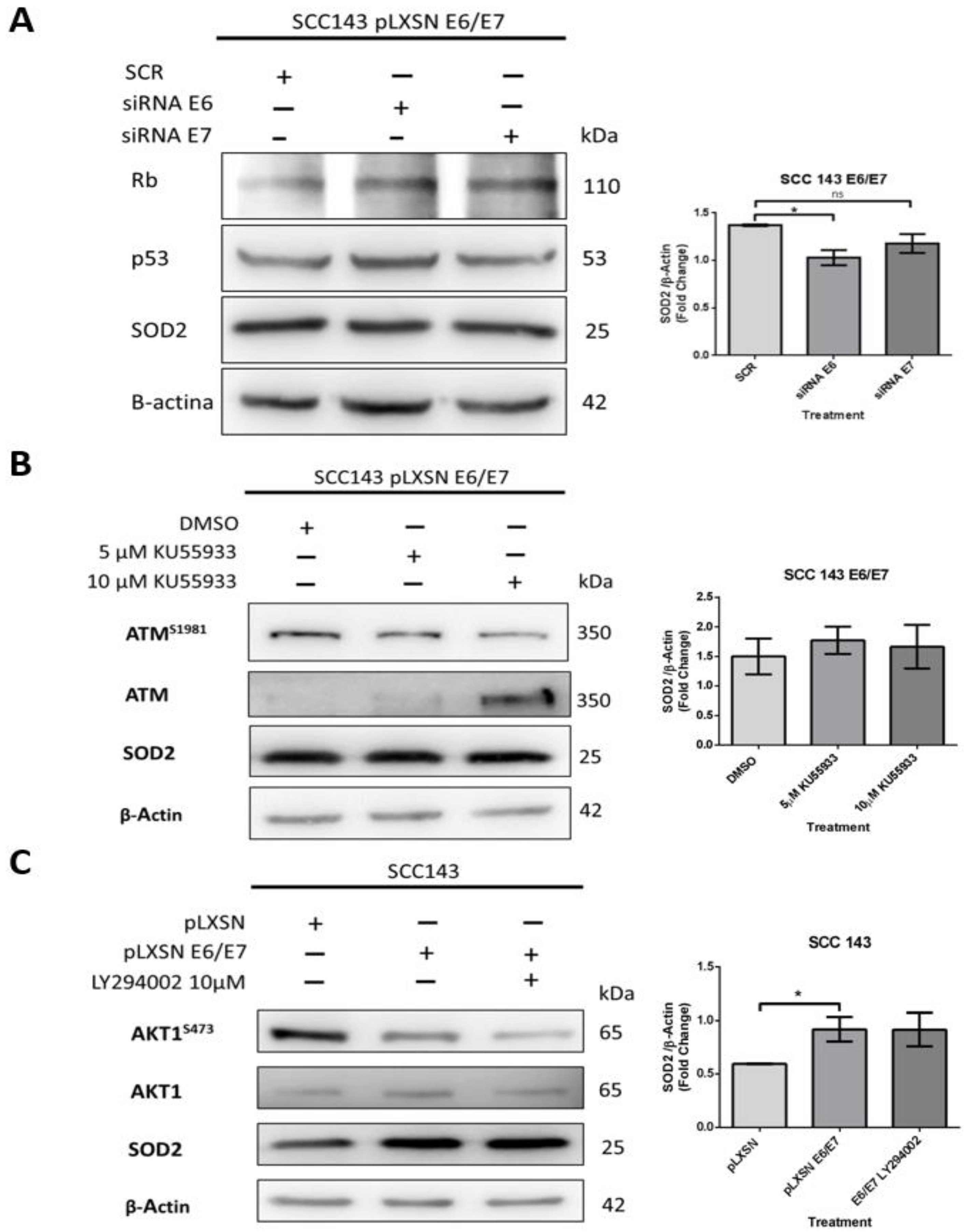
2.2. SOD2 Is Regulated by HPV16 E6 in an AKT1/ATM Independent Manner in Oral Cells
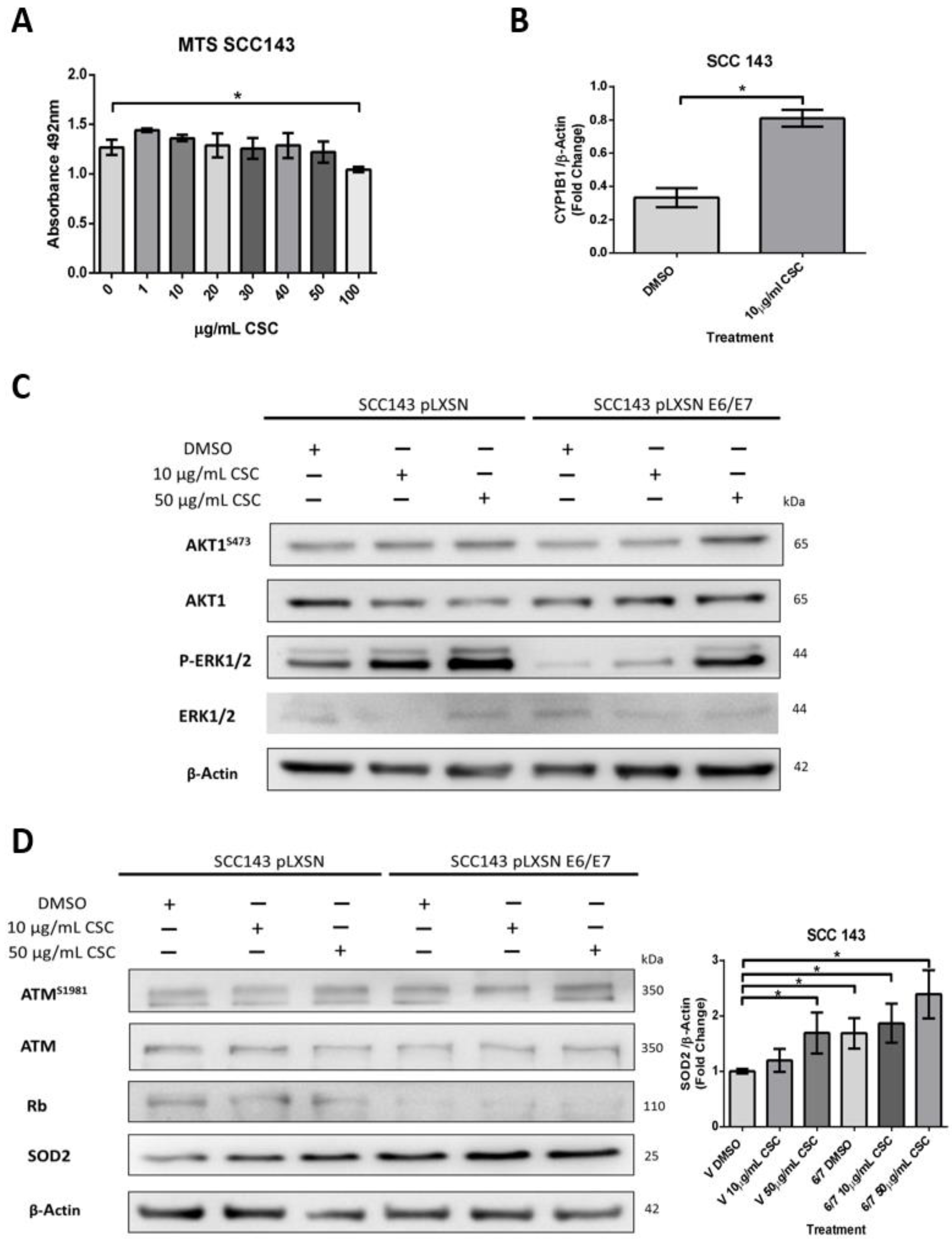
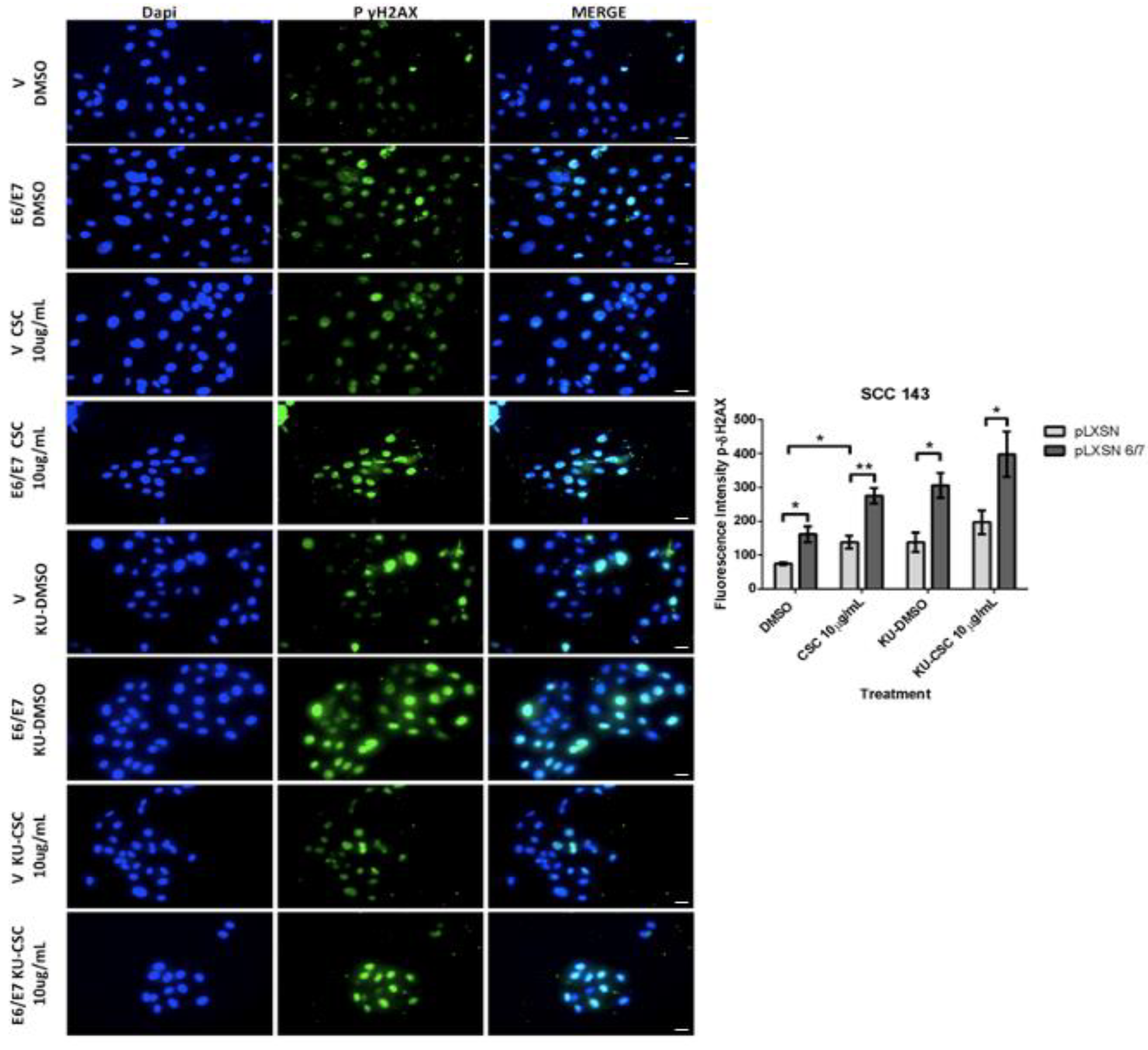
2.3. HPV16 E6 and E7 Oncoproteins Together with CSC Induce an Increase in SOD2 Levels and DNA Damage in Oral Cells
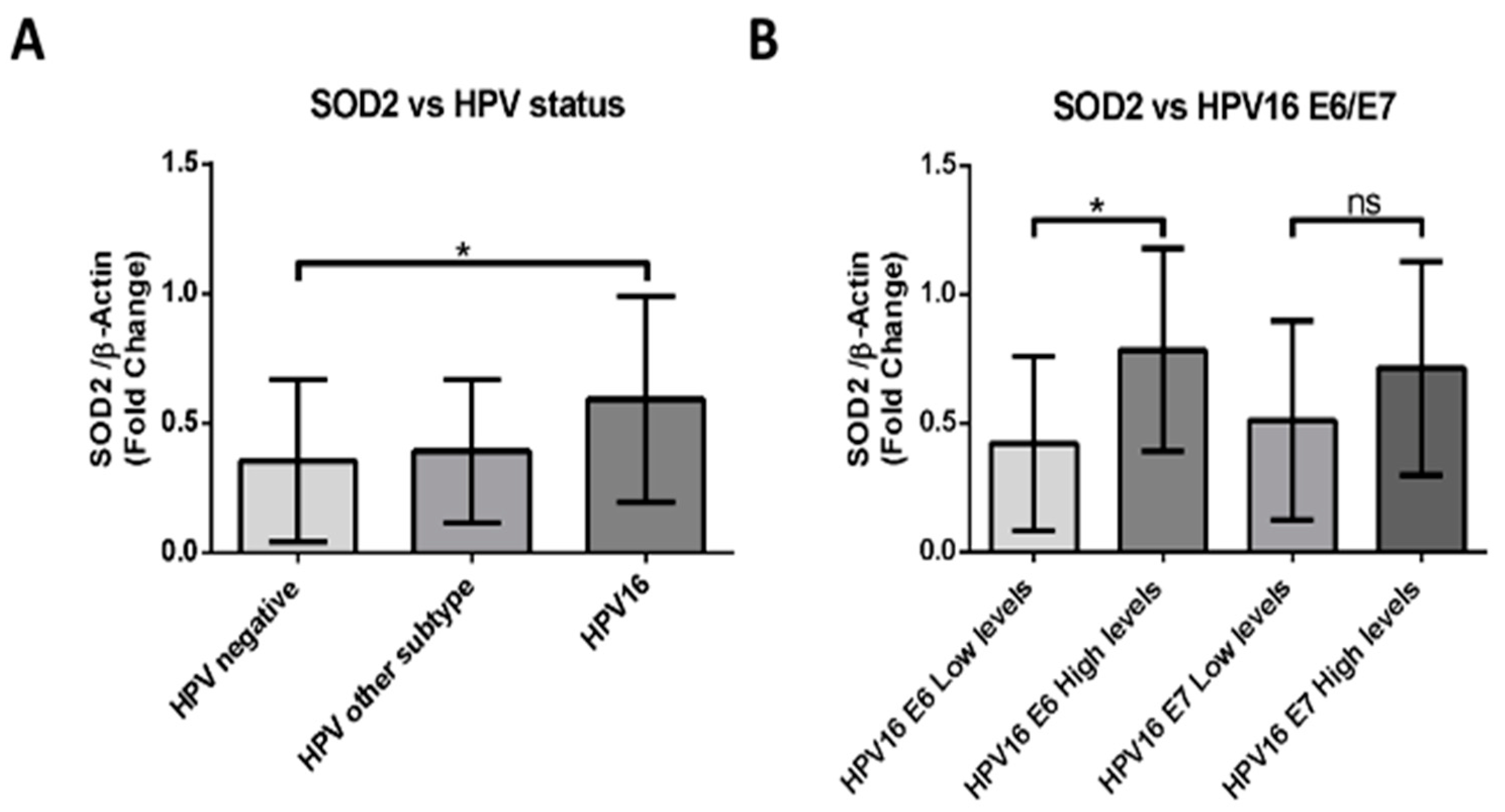
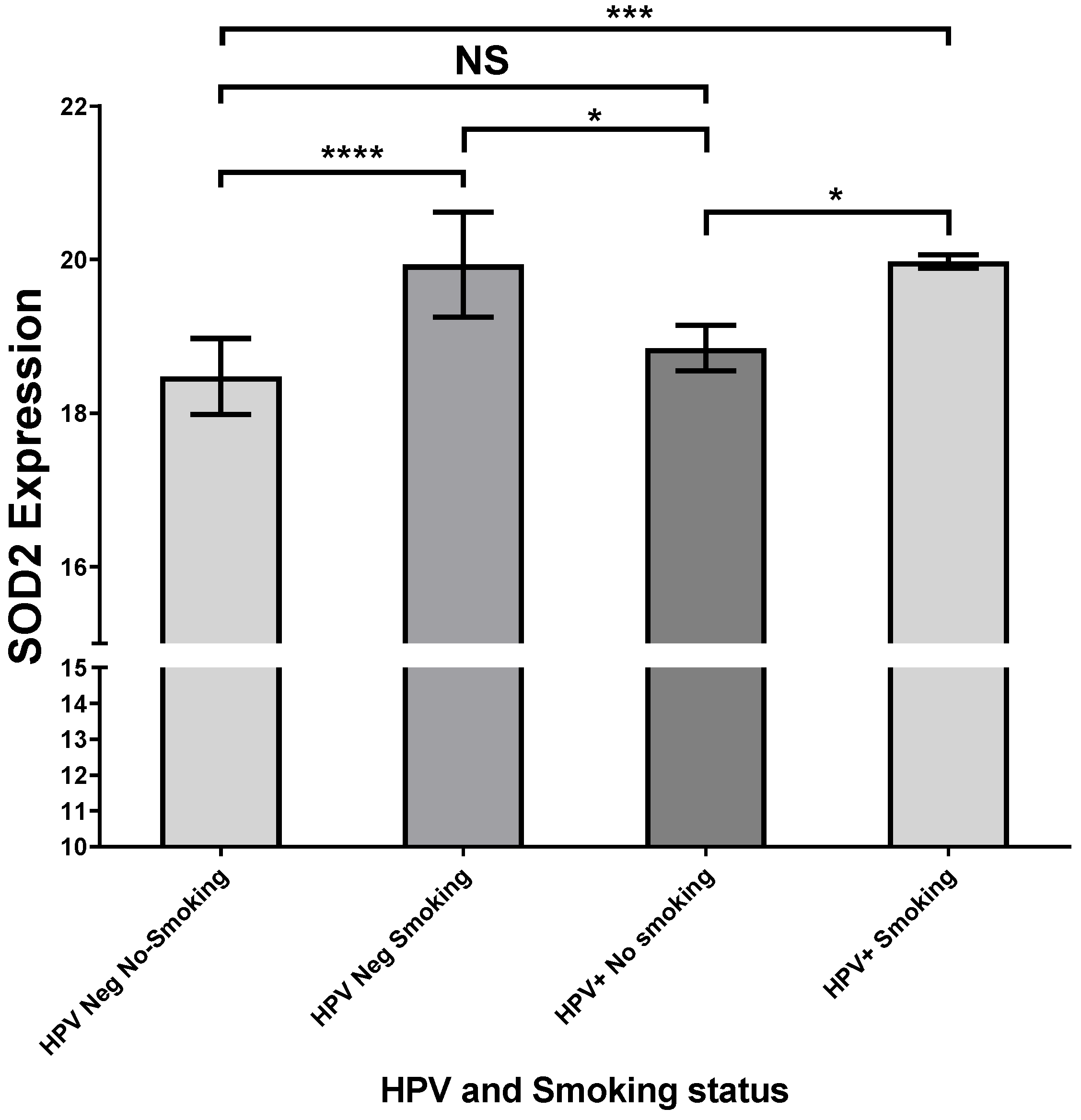
2.4. HPV16-Positive HNSCCs Correlate with Upregulation of SOD2 Transcripts
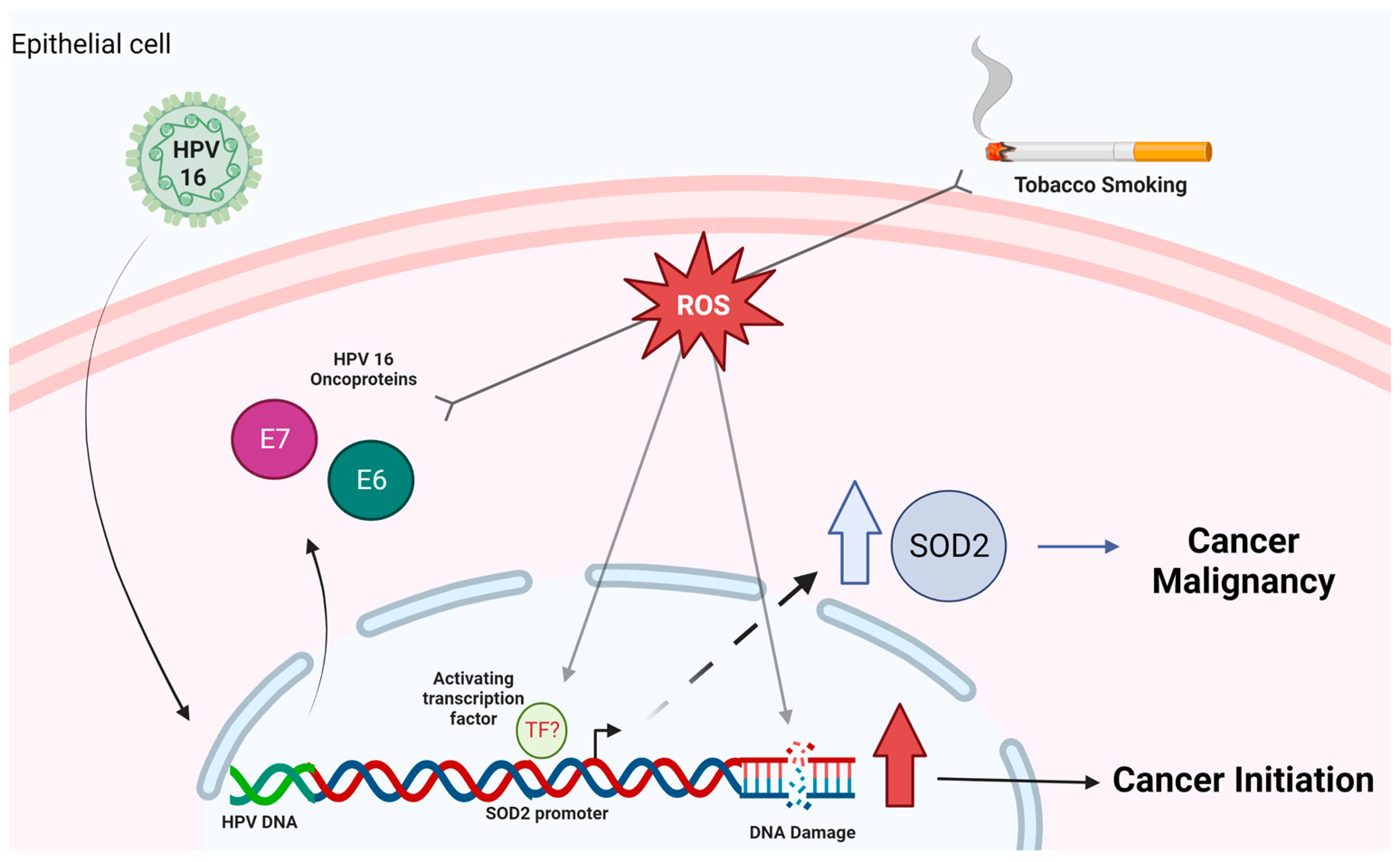
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture and Transductions
4.2. Viability Assays (MTS)
4.3. Real-Time Polymerase Chain Reaction (qPCR)
4.4. Western Blot
4.5. Immunofluorescence
4.6. Tissue Samples
4.7. FFPE RNA Extraction, cDNA Conversion, and RT-PCR
4.8. Gene Expression Analysis and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Hsieh, C.Y.; Lin, C.C.; Huang, Y.W.; Chen, J.H.; Tsou, Y.A.; Chang, L.C.; Fan, C.C.; Lin, C.Y.; Chang, W.C. Macrophage secretory IL-1beta promotes docetaxel resistance in head and neck squamous carcinoma via SOD2/CAT-ICAM1 signaling. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Heck, D.V.; Yee, C.L.; Howley, P.M.; Munger, K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc. Natl. Acad. Sci. USA 1992, 89, 4442–4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumban, E. A Current Update on Human Papillomavirus-Associated Head and Neck Cancers. Viruses 2019, 11, 922. [Google Scholar] [CrossRef] [Green Version]
- Reyes, M.; Rojas-Alcayaga, G.; Pennacchiotti, G.; Carrillo, D.; Munoz, J.P.; Pena, N.; Montes, R.; Lobos, N.; Aguayo, F. Human papillomavirus infection in oral squamous cell carcinomas from Chilean patients. Exp. Mol. Pathol. 2015, 99, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Hubbers, C.U.; Akgul, B. HPV and cancer of the oral cavity. Virulence 2015, 6, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.P.; Gonzalez, C.; Parra, B.; Corvalan, A.H.; Tornesello, M.L.; Eizuru, Y.; Aguayo, F. Functional interaction between human papillomavirus type 16 E6 and E7 oncoproteins and cigarette smoke components in lung epithelial cells. PLoS ONE 2012, 7, e38178. [Google Scholar] [CrossRef]
- Chen, X.; Mims, J.; Huang, X.; Singh, N.; Motea, E.; Planchon, S.M.; Beg, M.; Tsang, A.W.; Porosnicu, M.; Kemp, M.L.; et al. Modulators of Redox Metabolism in Head and Neck Cancer. Antioxid. Redox Signal. 2018, 29, 1660–1690. [Google Scholar] [CrossRef]
- Tang, M.S.; Lee, H.W.; Weng, M.W.; Wang, H.T.; Hu, Y.; Chen, L.C.; Park, S.H.; Chan, H.W.; Xu, J.; Wu, X.R.; et al. DNA damage, DNA repair and carcinogenicity: Tobacco smoke versus electronic cigarette aerosol. Mutat. Res./Rev. Mutat. Res. 2022, 789, 108409. [Google Scholar] [CrossRef] [PubMed]
- Aguayo, F.; Munoz, J.P.; Perez-Dominguez, F.; Carrillo-Beltran, D.; Oliva, C.; Calaf, G.M.; Blanco, R.; Nunez-Acurio, D. High-Risk Human Papillomavirus and Tobacco Smoke Interactions in Epithelial Carcinogenesis. Cancers 2020, 12, 2201. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Gold, L.S. Endogenous mutagens and the causes of aging and cancer. Mutat. Res. 1991, 250, 3–16. [Google Scholar] [CrossRef] [Green Version]
- De Marco, F. Oxidative stress and HPV carcinogenesis. Viruses 2013, 5, 708–731. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Jiang, R.S.; Wu, S.H.; Chen, F.J.; Liu, S.A. Smoking, alcohol, and betel quid and oral cancer: A prospective cohort study. J. Oncol. 2011, 2011, 525976. [Google Scholar] [CrossRef]
- Carrillo, D.; Munoz, J.P.; Huerta, H.; Leal, G.; Corvalan, A.; Leon, O.; Calaf, G.M.; Urzua, U.; Boccardo, E.; Tapia, J.C.; et al. Upregulation of PIR gene expression induced by human papillomavirus E6 and E7 in epithelial oral and cervical cells. Open Biol. 2017, 7, 170111. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Beltran, D.; Munoz, J.P.; Guerrero-Vasquez, N.; Blanco, R.; Leon, O.; de Souza Lino, V.; Tapia, J.C.; Maldonado, E.; Dubois-Camacho, K.; Hermoso, M.A.; et al. Human Papillomavirus 16 E7 Promotes EGFR/PI3K/AKT1/NRF2 Signaling Pathway Contributing to PIR/NF-kappaB Activation in Oral Cancer Cells. Cancers 2020, 12, 1904. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, H.; Sakimoto, M.; Yamashita, Y.; Murata, T.; Kajiyama, M. Manganese superoxide dismutase (Mn-SOD) correlates with prognosis of patients with oral squamous cell carcinoma. Fukuoka Igaku Zasshi 1998, 89, 321–327. [Google Scholar]
- Noh, J.K.; Woo, S.R.; Yun, M.; Lee, M.K.; Kong, M.; Min, S.; Kim, S.I.; Lee, Y.C.; Eun, Y.G.; Ko, S.G. SOD2- and NRF2-associated Gene Signature to Predict Radioresistance in Head and Neck Cancer. Cancer Genom. Proteom. 2021, 18, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Gupta Vallur, P.; Phaeton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants 2017, 6, 86. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.K.; Tangpong, J.; Chaiswing, L.; Oberley, T.D.; St Clair, D.K. Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages. Cancer Res. 2011, 71, 6684–6695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, K.M.; Hempel, N.; Nelson, K.K.; Dabiri, G.; Gamarra, A.; Belarmino, J.; Van De Water, L.; Mian, B.M.; Melendez, J.A. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Res. 2007, 67, 10260–10267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Li, S.; Cai, Y.; Wang, A.; He, Q.; Zheng, C.; Zhao, T.; Ding, X.; Zhou, X. Manganese superoxide dismutase induces migration and invasion of tongue squamous cell carcinoma via H2O2-dependent Snail signaling. Free Radic. Biol. Med. 2012, 53, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Talarico, M.C.R.; Nunes, R.A.L.; Silva, G.A.F.; Costa, L.; Cardoso, M.R.; Esteves, S.C.B.; Zanatta Sarian, L.O.; Zeferino, L.C.; Termini, L. High Expression of SOD2 Protein Is a Strong Prognostic Factor for Stage IIIB Squamous Cell Cervical Carcinoma. Antioxidants 2021, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Lee, H.M.; Kim, J.H.; Park, I.S.; Rho, Y.S. Heavy alcohol drinking downregulates ALDH2 gene expression but heavy smoking up-regulates SOD2 gene expression in head and neck squamous cell carcinoma. World J. Surg. Oncol. 2017, 15, 163. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Aparicio-Trejo, O.E.; Coronado-Martinez, I.; Pedraza-Chaverri, J.; Lizano, M. E6 Oncoproteins from High-Risk Human Papillomavirus Induce Mitochondrial Metabolism in a Head and Neck Squamous Cell Carcinoma Model. Biomolecules 2019, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [Green Version]
- Sobus, S.L.; Warren, G.W. The biologic effects of cigarette smoke on cancer cells. Cancer 2014, 120, 3617–3626. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2019, 10, 3116. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; D’Souza, G.; Westra, W.; Sugar, E.; Xiao, W.; Begum, S.; Viscidi, R. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J. Natl. Cancer Inst. 2008, 100, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Lerman, M.A.; Almazrooa, S.; Lindeman, N.; Hall, D.; Villa, A.; Woo, S.B. HPV-16 in a distinct subset of oral epithelial dysplasia. Mod. Pathol. 2017, 30, 1646–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, B.L.; Dierks, E.J.; Homer, L.; Potter, B. Tobacco smoking history and presentation of oral squamous cell carcinoma. J. Oral Maxillofac. Surg. 2004, 62, 1055–1058. [Google Scholar] [CrossRef]
- Smith, E.M.; Rubenstein, L.M.; Haugen, T.H.; Hamsikova, E.; Turek, L.P. Tobacco and alcohol use increases the risk of both HPV-associated and HPV-independent head and neck cancers. Cancer Causes Control 2010, 21, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Wittekindt, C.; Wagner, S.; Sharma, S.J.; Wurdemann, N.; Knuth, J.; Reder, H.; Klussmann, J.P. HPV—A different view on Head and Neck Cancer. Laryngo-Rhino-Otologie 2018, 97, S48–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurdemann, N.; Wagner, S.; Sharma, S.J.; Prigge, E.S.; Reuschenbach, M.; Gattenlohner, S.; Klussmann, J.P.; Wittekindt, C. Prognostic Impact of AJCC/UICC 8th Edition New Staging Rules in Oropharyngeal Squamous Cell Carcinoma. Front. Oncol. 2017, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Saraiya, M.; Unger, E.R.; Thompson, T.D.; Lynch, C.F.; Hernandez, B.Y.; Lyu, C.W.; Steinau, M.; Watson, M.; Wilkinson, E.J.; Hopenhayn, C.; et al. US assessment of HPV types in cancers: Implications for current and 9-valent HPV vaccines. J. Natl. Cancer Inst. 2015, 107, djv086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, C.; Carrillo-Beltran, D.; Boettiger, P.; Gallegos, I.; Aguayo, F. Human Papillomavirus Detected in Oropharyngeal Cancers from Chilean Subjects. Viruses 2022, 14, 1212. [Google Scholar] [CrossRef]
- Michaud, D.S.; Langevin, S.M.; Eliot, M.; Nelson, H.H.; Pawlita, M.; McClean, M.D.; Kelsey, K.T. High-risk HPV types and head and neck cancer. Int. J. Cancer 2014, 135, 1653–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, A.; Lo Muzio, L.; Kolokythas, A.; Sheng, S.; Rubini, C.; Ye, H.; Shi, F.; Yu, T.; Crowe, D.L.; et al. Deregulation of manganese superoxide dismutase (SOD2) expression and lymph node metastasis in tongue squamous cell carcinoma. BMC Cancer 2010, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Cao, C.; Huang, Z.; Tang, D.; Chen, J.; Wang, A.; He, Q. SOD2 confers anlotinib resistance via regulation of mitochondrial damage in OSCC. Oral Dis. 2022. [Google Scholar] [CrossRef]
- Jung, C.H.; Kim, E.M.; Song, J.Y.; Park, J.K.; Um, H.D. Mitochondrial superoxide dismutase 2 mediates gamma-irradiation-induced cancer cell invasion. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, M.; Choi, A.J.; Lee, Y.C.; Kong, M.; Sung, J.Y.; Kim, S.S.; Eun, Y.G. Carbonyl reductase 1 is a new target to improve the effect of radiotherapy on head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 264. [Google Scholar] [CrossRef]
- Termini, L.; Filho, A.L.; Maciag, P.C.; Etlinger, D.; Alves, V.A.; Nonogaki, S.; Soares, F.A.; Villa, L.L. Deregulated expression of superoxide dismutase-2 correlates with different stages of cervical neoplasia. Dis. Markers 2011, 30, 275–281. [Google Scholar] [CrossRef]
- Rabelo-Santos, S.H.; Termini, L.; Boccardo, E.; Derchain, S.; Longatto-Filho, A.; Andreoli, M.A.; Costa, M.C.; Lima Nunes, R.A.; Lucci Angelo-Andrade, L.A.; Villa, L.L.; et al. Strong SOD2 expression and HPV-16/18 positivity are independent events in cervical cancer. Oncotarget 2018, 9, 21630–21640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, L.M.; Kepple, J.D.; Ai, L.; Kim, W.J.; Stanton, V.L.; Reinhard, M.K.; Backman, L.R.F.; Streitfeld, W.S.; Babu, N.R.; Treiber, N.; et al. ATM is required for SOD2 expression and homeostasis within the mammary gland. Breast Cancer Res. Treat. 2017, 166, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carloni, S.; Girelli, S.; Scopa, C.; Buonocore, G.; Longini, M.; Balduini, W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010, 6, 366–377. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, V.; Iommelli, F.; Monti, M.; Fonti, R.; Votta, G.; Stoppelli, M.P.; Del Vecchio, S. Reversal of Warburg Effect and Reactivation of Oxidative Phosphorylation by Differential Inhibition of EGFR Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 5110–5120. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Porntadavity, S.; St Clair, D.K. Transcriptional regulation of the human manganese superoxide dismutase gene: The role of specificity protein 1 (Sp1) and activating protein-2 (AP-2). Biochem. J. 2002, 362 Pt 2, 401–412. [Google Scholar] [CrossRef]
- Gunnell, A.S.; Tran, T.N.; Torrang, A.; Dickman, P.W.; Sparen, P.; Palmgren, J.; Ylitalo, N. Synergy between cigarette smoking and human papillomavirus type 16 in cervical cancer in situ development. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2141–2147. [Google Scholar] [CrossRef] [Green Version]
- Louie, K.S.; Castellsague, X.; de Sanjose, S.; Herrero, R.; Meijer, C.J.; Shah, K.; Munoz, N.; Bosch, F.X. Smoking and passive smoking in cervical cancer risk: Pooled analysis of couples from the IARC multicentric case-control studies. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Pena, N.; Carrillo, D.; Munoz, J.P.; Chnaiderman, J.; Urzua, U.; Leon, O.; Tornesello, M.L.; Corvalan, A.H.; Soto-Rifo, R.; Aguayo, F. Tobacco smoke activates human papillomavirus 16 p97 promoter and cooperates with high-risk E6/E7 for oxidative DNA damage in lung cells. PLoS ONE 2015, 10, e0123029. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.P.; Carrillo-Beltran, D.; Aedo-Aguilera, V.; Calaf, G.M.; Leon, O.; Maldonado, E.; Tapia, J.C.; Boccardo, E.; Ozbun, M.A.; Aguayo, F. Tobacco Exposure Enhances Human Papillomavirus 16 Oncogene Expression via EGFR/PI3K/Akt/c-Jun Signaling Pathway in Cervical Cancer Cells. Front. Microbiol. 2018, 9, 3022. [Google Scholar] [CrossRef]
- Ndisang, D.; Khan, A.; Lorenzato, F.; Sindos, M.; Singer, A.; Latchman, D.S. The cellular transcription factor Brn-3a and the smoking-related substance nicotine interact to regulate the activity of the HPV URR in the cervix. Oncogene 2010, 29, 2701–2711. [Google Scholar] [CrossRef] [Green Version]
- Koshiol, J.; Schroeder, J.; Jamieson, D.J.; Marshall, S.W.; Duerr, A.; Heilig, C.M.; Shah, K.V.; Klein, R.S.; Cu-Uvin, S.; Schuman, P.; et al. Smoking and time to clearance of human papillomavirus infection in HIV-seropositive and HIV-seronegative women. Am. J. Epidemiol. 2006, 164, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Trushin, N.; Alam, S.; El-Bayoumy, K.; Krzeminski, J.; Amin, S.G.; Gullett, J.; Meyers, C.; Prokopczyk, B. Comparative metabolism of benzo[a]pyrene by human keratinocytes infected with high-risk human papillomavirus types 16 and 18 as episomal or integrated genomes. J. Carcinog. 2012, 11, 1. [Google Scholar] [CrossRef]
- Moktar, A.; Ravoori, S.; Vadhanam, M.V.; Gairola, C.G.; Gupta, R.C. Cigarette smoke-induced DNA damage and repair detected by the comet assay in HPV-transformed cervical cells. Int. J. Oncol. 2009, 35, 1297–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, H. eIF4E is a critical regulator of human papillomavirus (HPV)-immortalized cervical epithelial (H8) cell growth induced by nicotine. Toxicology 2019, 419, 1–10. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhalawani, H.; Mohamed, A.S.R.; Elgohari, B.; Lin, T.A.; Sikora, A.G.; Lai, S.Y.; Abusaif, A.; Phan, J.; Morrison, W.H.; Gunn, G.B.; et al. Tobacco exposure as a major modifier of oncologic outcomes in human papillomavirus (HPV) associated oropharyngeal squamous cell carcinoma. BMC Cancer 2020, 20, 912. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, C.; Gillison, M.L.; D’Souza, G. Tobacco use and oral HPV-16 infection. JAMA 2014, 312, 1465–1467. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, S.Y.; Broglie, M.A.; Soltermann, A.; Huber, G.F.; Stoeckli, S.J. Comparison of PI3K Pathway in HPV-Associated Oropharyngeal Cancer with and without Tobacco Exposure. Laryngoscope Investig. Otolaryngol. 2018, 3, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S.; Itani, H.; Richmond, B.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; Dikalova, A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H639–H646. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Rabinovitch, P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy 2011, 7, 917–918. [Google Scholar] [CrossRef] [Green Version]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Lin, Z.; Pavlides, S.; Whitaker-Menezes, D.; Pestell, R.G.; Howell, A.; Sotgia, F. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: The seed and soil also needs “fertilizer”. Cell Cycle 2011, 10, 2440–2449. [Google Scholar] [CrossRef] [Green Version]
- White, J.S.; Weissfeld, J.L.; Ragin, C.C.; Rossie, K.M.; Martin, C.L.; Shuster, M.; Ishwad, C.S.; Law, J.C.; Myers, E.N.; Johnson, J.T.; et al. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol. 2007, 43, 701–712. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward 5′-3′ | Reverse 5′-3′ | Size (bp) |
|---|---|---|---|
| E6 small 16 | CTGCAAGCAACAGTTACTGCGA | TCACACACTGCATATGGATTCCC | 96 |
| E7 small 16 | CAATATTGTAATGGGCTCTGTCC | ATTTGCAACCAGAGACAACTGAT | 120 |
| PCO3/PCO4 | ACACAACTGTGTTCACTAG | CAACTTCATCCACGTTCACC | 110 |
| GP5+/GP6+ | TTTGTTACTGTGGTAGATATCAC | GAAAAATAAACTTAAATCATATTC | 155 |
| SOD2 | GCCCTGGAACCTCACATCAAC | CAACGCCTCCTGGTACTTCTC | 111 |
| b-actin | CCACACAGGGGAGGTGATAG | CCACACAGGGGAGGTGATAG | 115 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrillo-Beltrán, D.; Osorio, J.C.; Blanco, R.; Oliva, C.; Boccardo, E.; Aguayo, F. Interaction between Cigarette Smoke and Human Papillomavirus 16 E6/E7 Oncoproteins to Induce SOD2 Expression and DNA Damage in Head and Neck Cancer. Int. J. Mol. Sci. 2023, 24, 6907. https://doi.org/10.3390/ijms24086907
Carrillo-Beltrán D, Osorio JC, Blanco R, Oliva C, Boccardo E, Aguayo F. Interaction between Cigarette Smoke and Human Papillomavirus 16 E6/E7 Oncoproteins to Induce SOD2 Expression and DNA Damage in Head and Neck Cancer. International Journal of Molecular Sciences. 2023; 24(8):6907. https://doi.org/10.3390/ijms24086907
Chicago/Turabian StyleCarrillo-Beltrán, Diego, Julio C. Osorio, Rancés Blanco, Carolina Oliva, Enrique Boccardo, and Francisco Aguayo. 2023. "Interaction between Cigarette Smoke and Human Papillomavirus 16 E6/E7 Oncoproteins to Induce SOD2 Expression and DNA Damage in Head and Neck Cancer" International Journal of Molecular Sciences 24, no. 8: 6907. https://doi.org/10.3390/ijms24086907