
Folic Acid Ameliorates Renal Injury in Experimental Obstructive Nephropathy: Role of Glycine N-Methyltransferase
 , and
, and
Abstract
:1. Introduction
2. Results
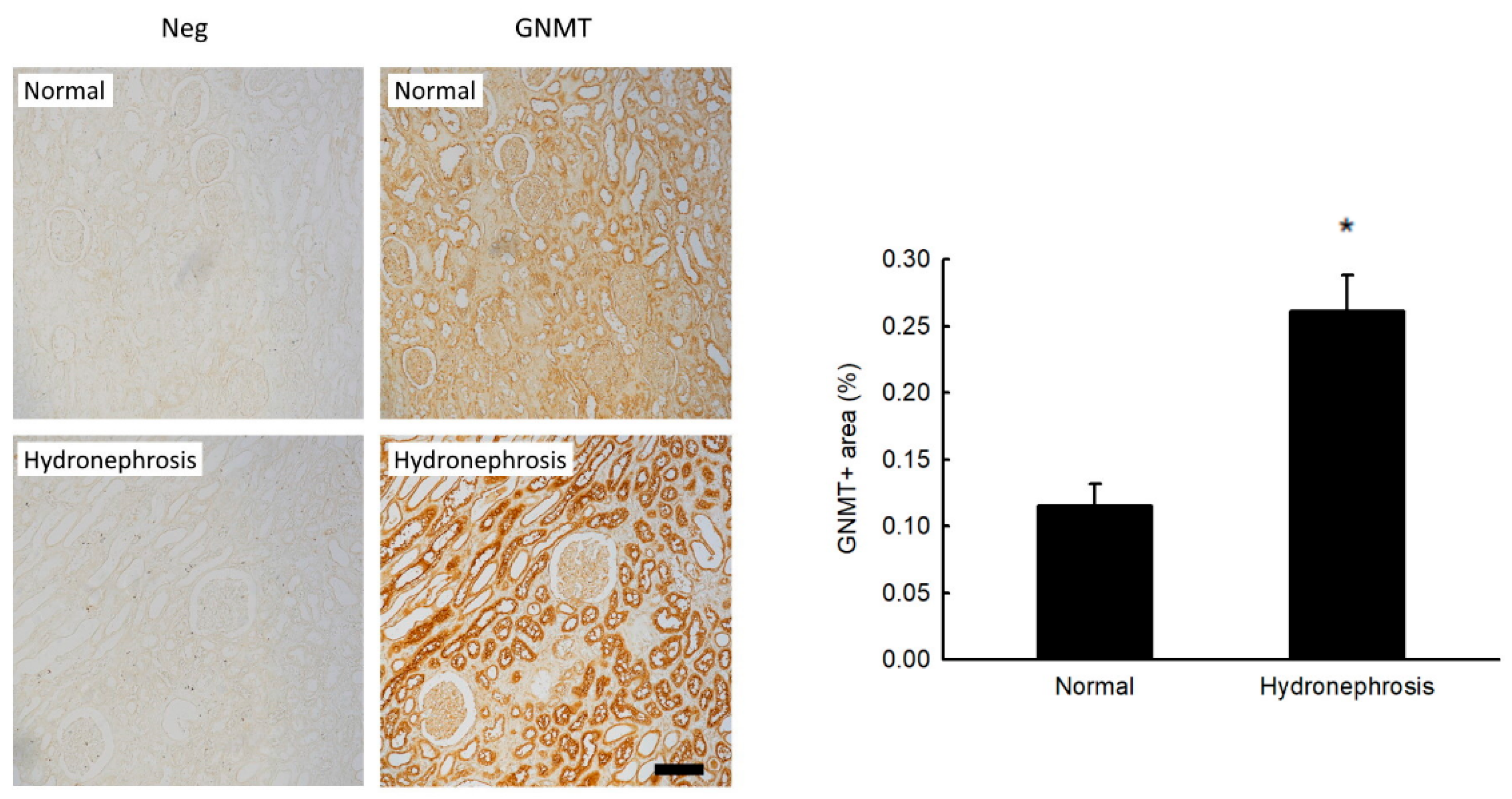
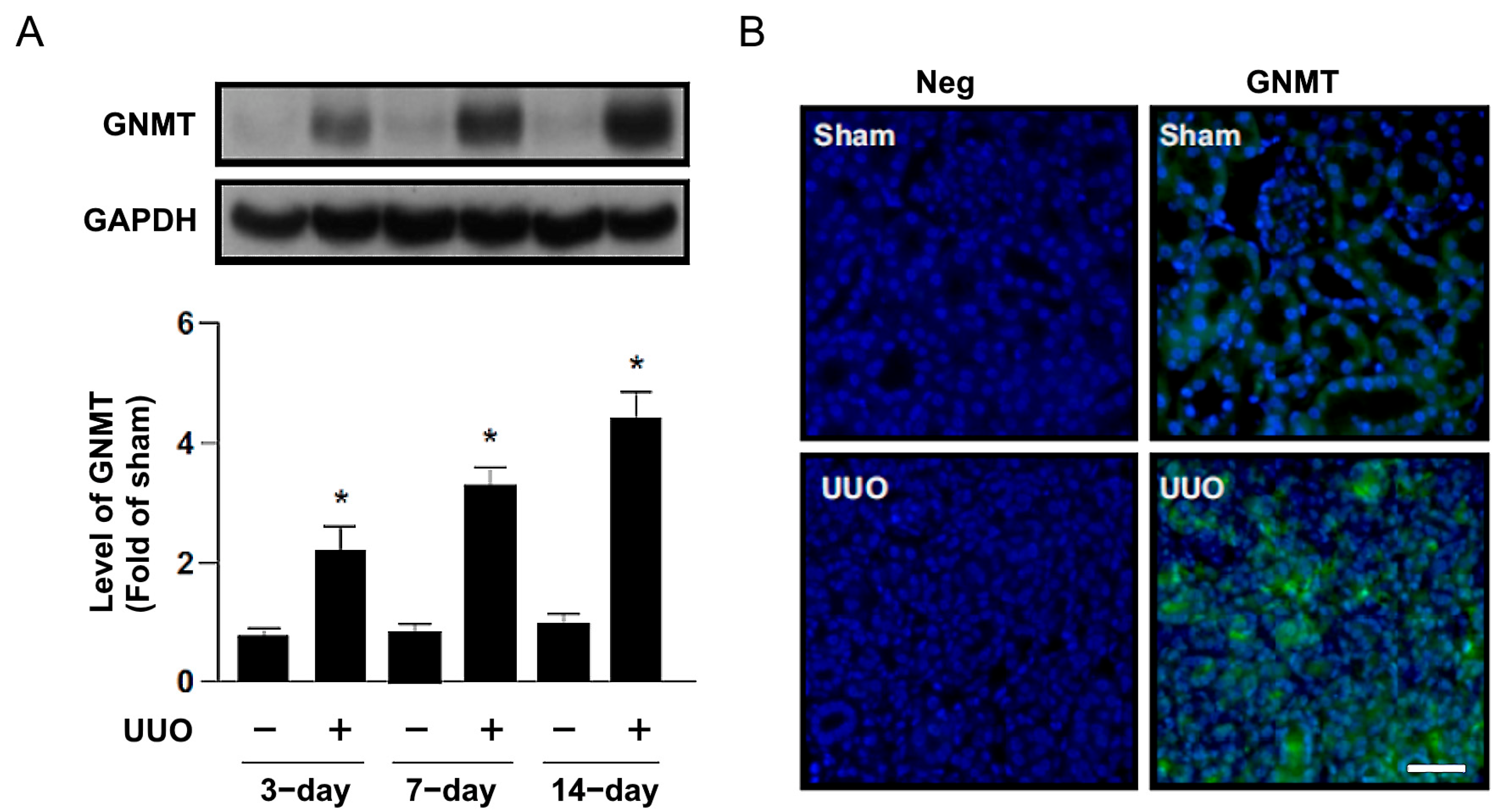
2.1. Increased GNMT Expression in Humans and Mice with Obstructive Nephropathy
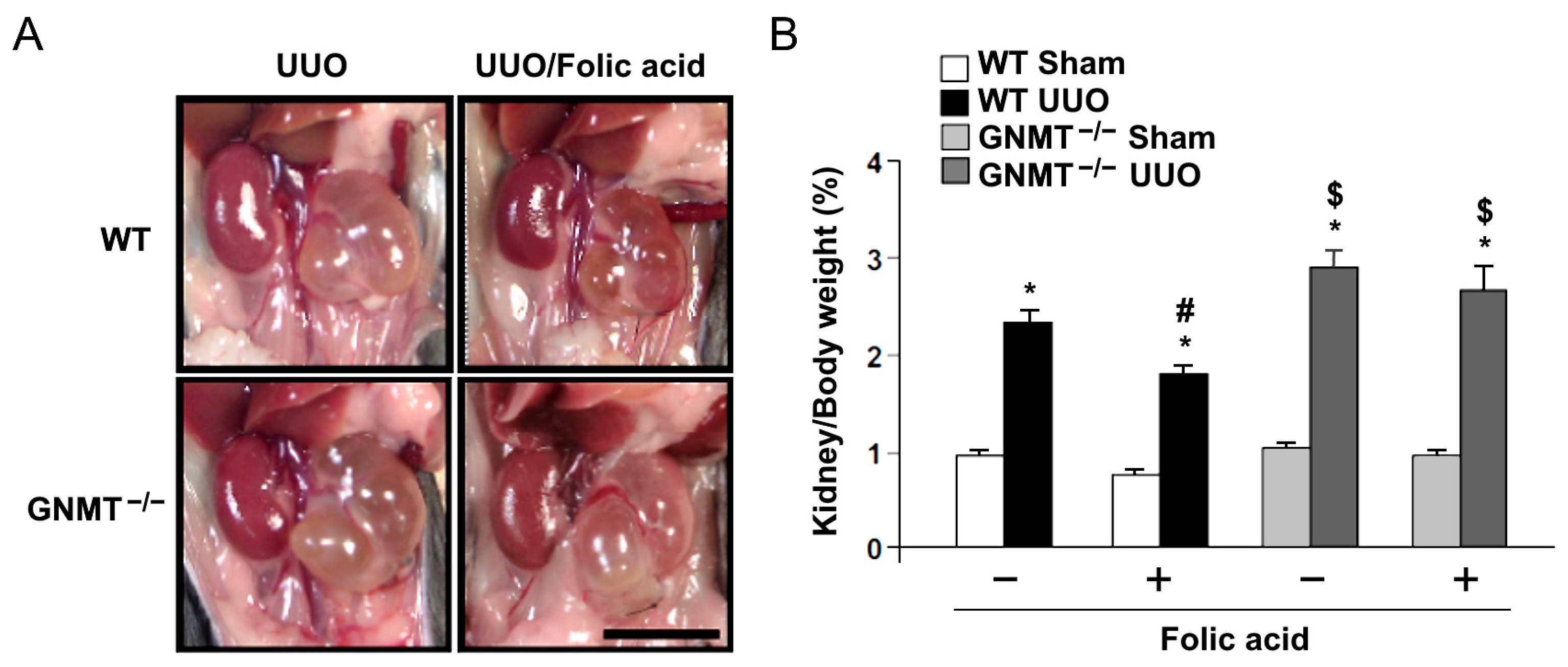
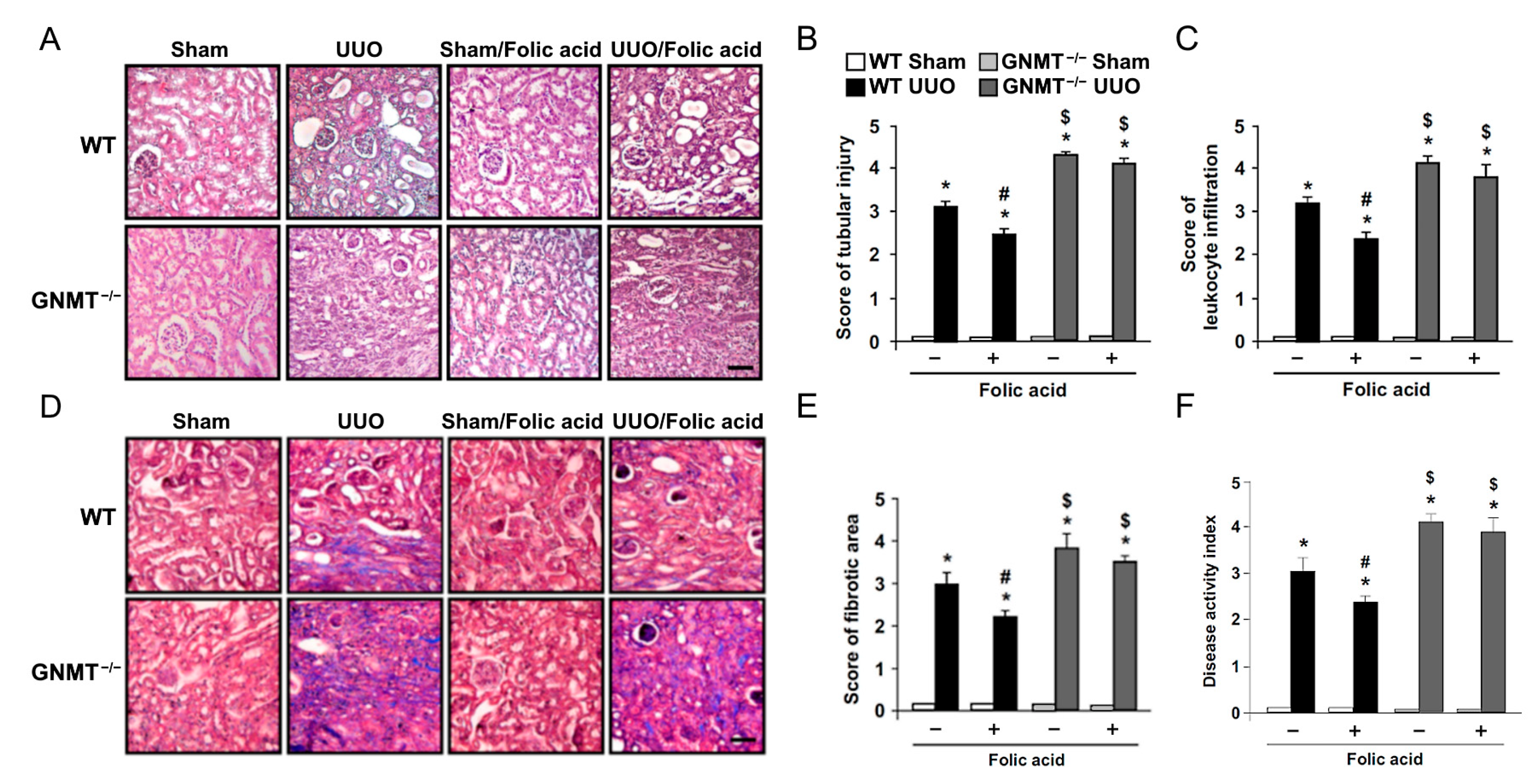
2.2. Treatment with Folic Acid Reduces Renal Injury in UUO Mice but Not in GNMT−/− Mice
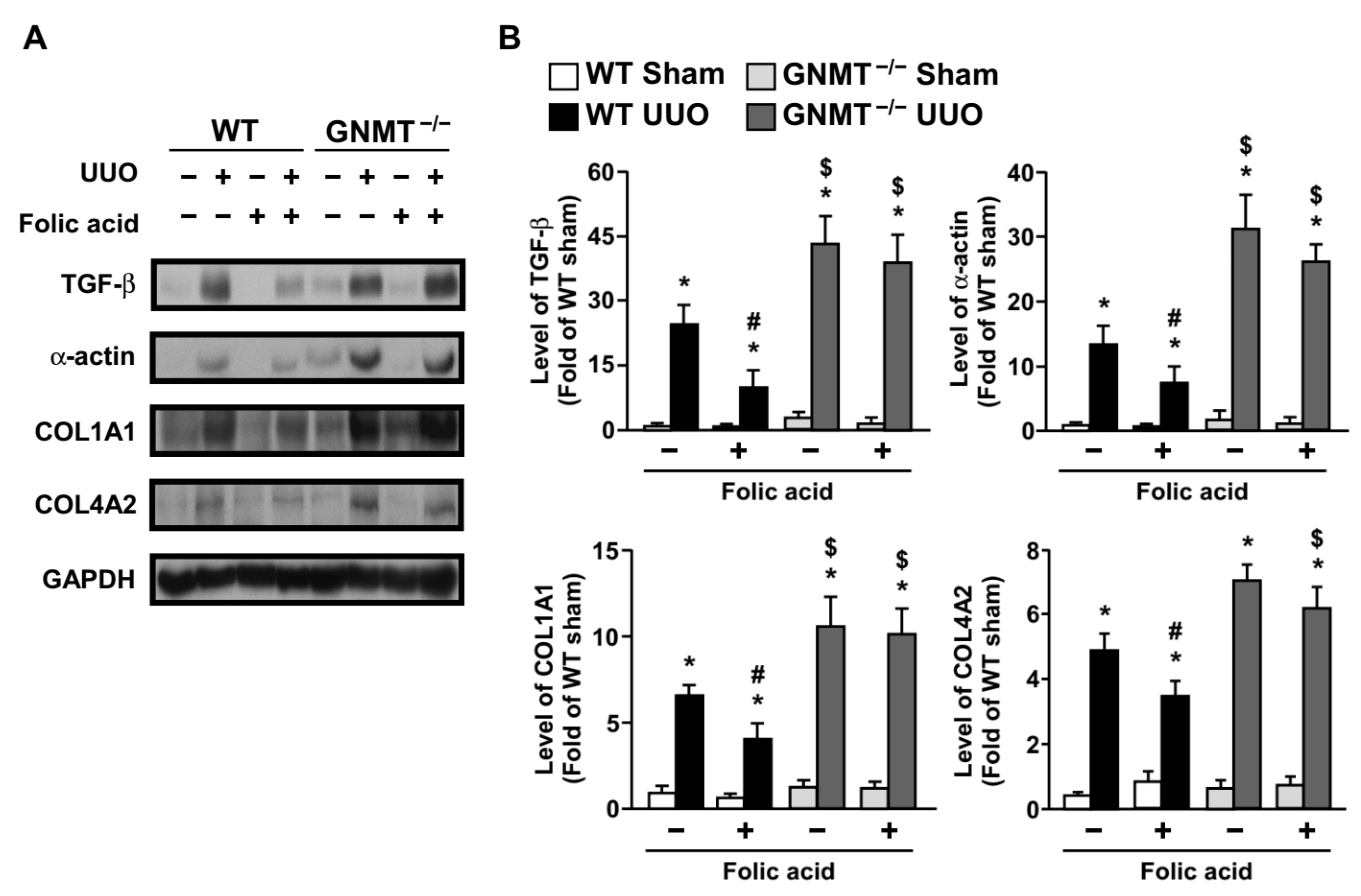
2.3. Treatment with Folic Acid Inhibits the Inflammatory Response and Decreases the Expression of Fibrosis-Related Proteins in UUO Mice but Not in GNMT−/− Mice
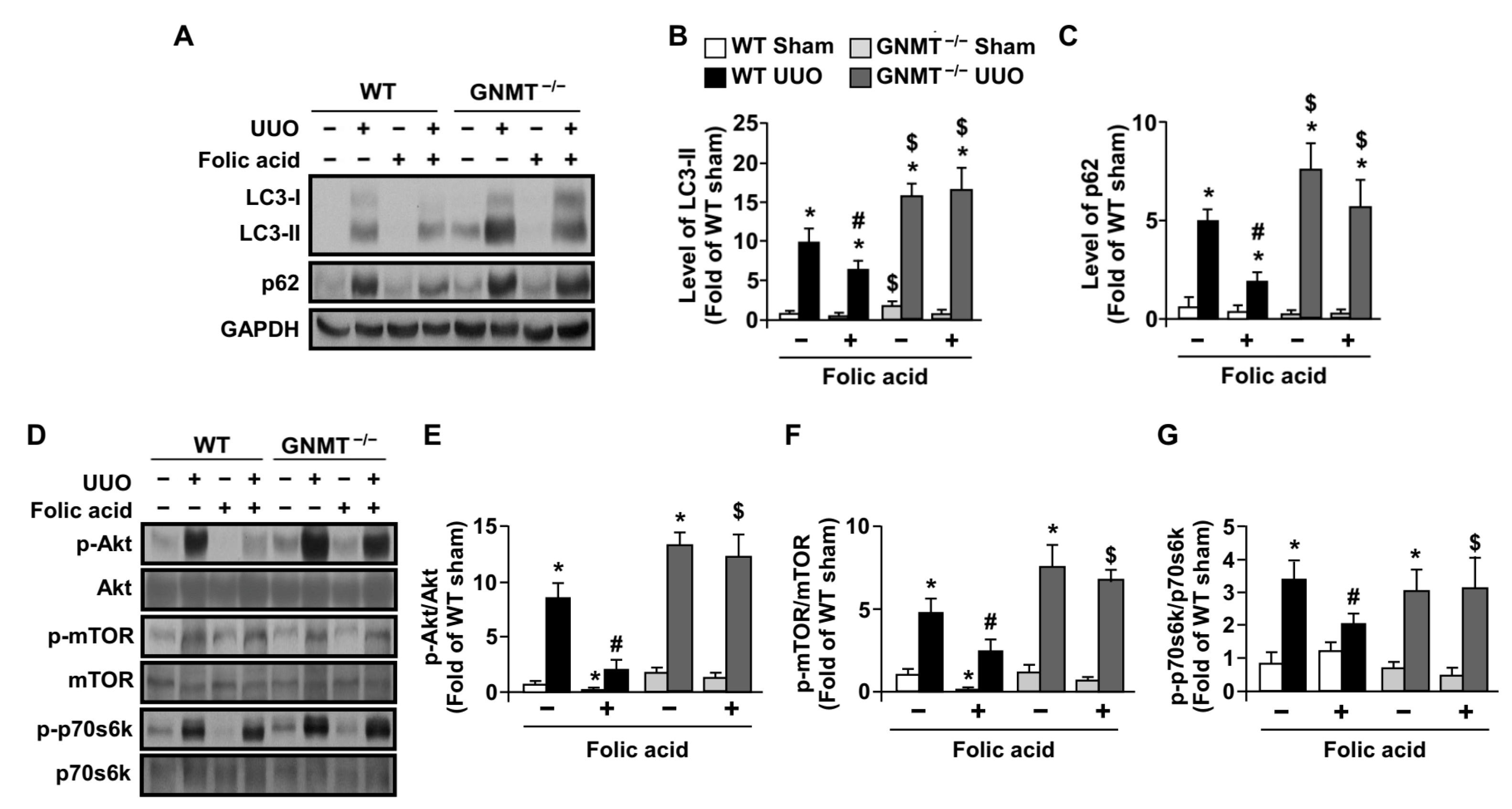
2.4. Treatment with Folic Acid Activates Autophagy Flux in UUO Mice but Not in GNMT−/− Mice
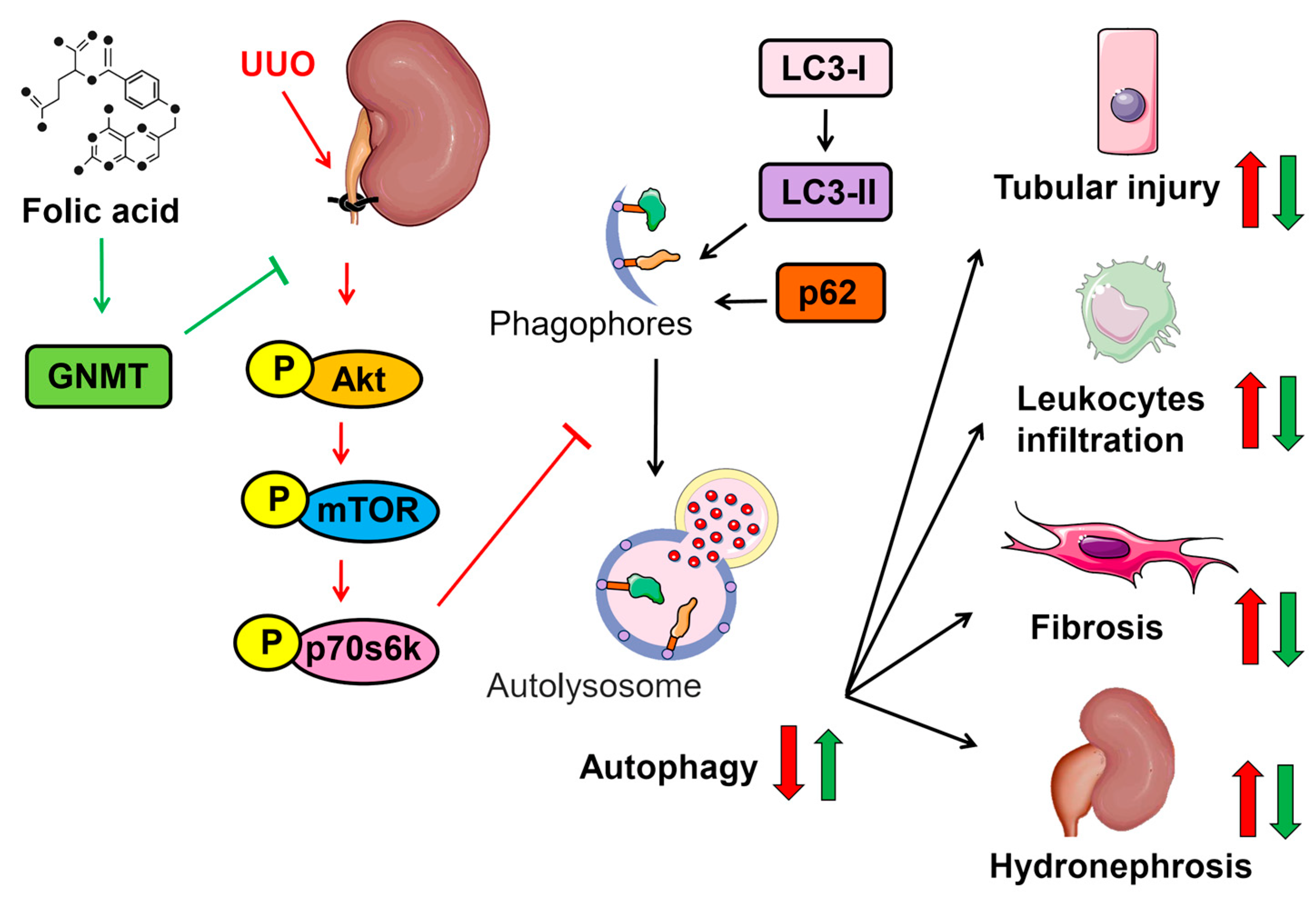
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Human Kidney Specimens and Experimental Animals
4.3. Unilateral Ureteral Obstruction (UUO) and Folic Acid Treatment
4.4. Immunohistochemical Analysis
4.5. Western Blot Analysis
4.6. Histological Examination
4.7. Masson’s Trichrome Staining
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klahr, S.; Morrissey, J. Obstructive nephropathy and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2002, 283, F861–F875. [Google Scholar] [CrossRef] [Green Version]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, L.; Bethunaickan, R.; Ramanujam, M.; Huang, W.; Schiffer, M.; Tao, H.; Madaio, M.P.; Bottinger, E.P.; Davidson, A. Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J. Immunol. 2008, 180, 1938–1947. [Google Scholar] [CrossRef] [Green Version]
- López-Novoa, J.M.; Martínez-Salgado, C.; Rodríguez-Peña, A.B.; López-Hernández, F.J. Common pathophysiological mechanisms of chronic kidney disease: Therapeutic perspectives. Pharmacol. Ther. 2010, 128, 61–81. [Google Scholar] [PubMed]
- Vielhauer, V.; Kulkarni, O.; Reichel, C.A.; Anders, H.J. Targeting the recruitment of monocytes and macrophages in renal disease. Semin. Nephrol. 2010, 30, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, G.F.; Harris, K.P.; Purkerson, M.L.; Klahr, S. Immunological aspects of acute ureteral obstruction: Immune cell infiltrate in the kidney. Kidney Int. 1988, 34, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Grande, M.T.; López-Novoa, J.M. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 2009, 5, 319–328. [Google Scholar] [CrossRef]
- Duthie, S.J. Folic acid deficiency and cancer: Mechanisms of DNA instability. Br. Med. Bull. 1999, 55, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Huo, Y.; Langman, C.B.; Hou, F.; Chen, Y.; Matossian, D.; Xu, X.; Wang, X. Folic acid therapy and cardiovascular disease in esrd or advanced chronic kidney disease: A meta-analysis. Clin. J. Am. Soc. Nephrol. CJASN 2011, 6, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, C.M.; Spence, J.D. Folic acid supplementation and chronic kidney disease progression. Kidney Int. 2016, 90, 1144–1145. [Google Scholar] [CrossRef]
- Xu, X.; Qin, X.; Li, Y.; Sun, D.; Wang, J.; Liang, M.; Wang, B.; Huo, Y.; Hou, F.F. Efficacy of folic acid therapy on the progression of chronic kidney disease: The renal substudy of the china stroke primary prevention trial. JAMA Intern. Med. 2016, 176, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.A.; Kim, Y.I.; Salas, M.C.; Gamble, M.V.; Shibata, W.; Muthupalani, S.; Sohn, K.J.; Abrams, J.A.; Fox, J.G.; Wang, T.C.; et al. Folic acid increases global DNA methylation and reduces inflammation to prevent helicobacter-associated gastric cancer in mice. Gastroenterology 2012, 142, 824–833.e827. [Google Scholar] [CrossRef] [PubMed]
- Qipshidze, N.; Tyagi, N.; Metreveli, N.; Lominadze, D.; Tyagi, S.C. Autophagy mechanism of right ventricular remodeling in murine model of pulmonary artery constriction. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H688–H696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebaid, H.; Bashandy, S.A.; Alhazza, I.M.; Rady, A.; El-Shehry, S. Folic acid and melatonin ameliorate carbon tetrachloride-induced hepatic injury, oxidative stress and inflammation in rats. Nutr. Metab. 2013, 10, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocardo, P.S.; Budni, J.; Kaster, M.P.; Santos, A.R.; Rodrigues, A.L. Folic acid administration produces an antidepressant-like effect in mice: Evidence for the involvement of the serotonergic and noradrenergic systems. Neuropharmacology 2008, 54, 464–473. [Google Scholar]
- Yeo, E.J.; Wagner, C. Tissue distribution of glycine n-methyltransferase, a major folate-binding protein of liver. Proc. Natl. Acad. Sci. USA 1994, 91, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Luka, Z.; Mudd, S.H.; Wagner, C. Glycine n-methyltransferase and regulation of s-adenosylmethionine levels. J. Biol. Chem. 2009, 284, 22507–22511. [Google Scholar] [CrossRef] [Green Version]
- DebRoy, S.; Kramarenko, I.I.; Ghose, S.; Oleinik, N.V.; Krupenko, S.A.; Krupenko, N.I. A novel tumor suppressor function of glycine n-methyltransferase is independent of its catalytic activity but requires nuclear localization. PLoS ONE 2013, 8, e70062. [Google Scholar]
- Liu, S.P.; Li, Y.S.; Chen, Y.J.; Chiang, E.P.; Li, A.F.; Lee, Y.H.; Tsai, T.F.; Hsiao, M.; Huang, S.F.; Chen, Y.M. Glycine n-methyltransferase−/− mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology 2007, 46, 1413–1425. [Google Scholar] [CrossRef]
- Chen, Y.M.; Shiu, J.Y.; Tzeng, S.J.; Shih, L.S.; Chen, Y.J.; Lui, W.Y.; Chen, P.H. Characterization of glycine-n-methyltransferase-gene expression in human hepatocellular carcinoma. Int. J. Cancer 1998, 75, 787–793. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; Vázquez-Chantada, M.; Ariz, U.; Martínez, N.; Varela, M.; Luka, Z.; Capdevila, A.; Rodríguez, J.; Aransay, A.M.; Matthiesen, R.; et al. Loss of the glycine n-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology 2008, 47, 1191–1199. [Google Scholar]
- Chen, C.Y.; Ching, L.C.; Liao, Y.J.; Yu, Y.B.; Tsou, C.Y.; Shyue, S.K.; Chen, Y.M.; Lee, T.S. Deficiency of glycine n-methyltransferase aggravates atherosclerosis in apolipoprotein e-null mice. Mol. Med. 2012, 18, 744–752. [Google Scholar] [PubMed]
- Chou, W.Y.; Zhao, J.F.; Chen, Y.M.; Lee, K.I.; Su, K.H.; Shyue, S.K.; Lee, T.S. Role of glycine n-methyltransferase in experimental ulcerative colitis. J. Gastroenterol. Hepatol. 2014, 29, 494–501. [Google Scholar] [PubMed]
- Cook, R.J.; Wagner, C. Glycine n-methyltransferase is a folate binding protein of rat liver cytosol. Proc. Natl. Acad. Sci. USA 1984, 81, 3631–3634. [Google Scholar]
- Wagner, C.; Briggs, W.T.; Cook, R.J. Inhibition of glycine n-methyltransferase activity by folate derivatives: Implications for regulation of methyl group metabolism. Biochem. Biophys. Res. Commun. 1985, 127, 746–752. [Google Scholar]
- Chiang, C.W.; Lee, H.T.; Tarng, D.C.; Kuo, K.L.; Cheng, L.C.; Lee, T.S. Genetic deletion of soluble epoxide hydrolase attenuates inflammation and fibrosis in experimental obstructive nephropathy. Mediat. Inflamm. 2015, 2015, 693260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, T.H.; Tseng, K.Y.; Tsao, W.S.; Yang, C.Y.; Hsieh, S.L.; Chiu, A.W.; Takai, T.; Mak, T.W.; Tarng, D.C.; Chen, N.J. Trem-1 regulates macrophage polarization in ureteral obstruction. Kidney Int. 2014, 86, 1174–1186. [Google Scholar]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar]
- Chevalier, R.L. Obstructive nephropathy: Towards biomarker discovery and gene therapy. Nat. Clin. Pract. Nephrol. 2006, 2, 157–168. [Google Scholar]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [PubMed] [Green Version]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [PubMed]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [PubMed] [Green Version]
- Zhang, Q.; Qiao, H.; Wu, D.; Lu, H.; Liu, L.; Sang, X.; Li, D.; Zhou, Y. Curcumin potentiates the galbanic acid-induced anti-tumor effect in non-small cell lung cancer cells through inhibiting akt/mtor signaling pathway. Life Sci. 2019, 239, 117044. [Google Scholar]
- Obata, F.; Kuranaga, E.; Tomioka, K.; Ming, M.; Takeishi, A.; Chen, C.H.; Soga, T.; Miura, M. Necrosis-driven systemic immune response alters sam metabolism through the foxo-gnmt axis. Cell Rep. 2014, 7, 821–833. [Google Scholar]
- Gomez-Santos, L.; Luka, Z.; Wagner, C.; Fernandez-Alvarez, S.; Lu, S.C.; Mato, J.M.; Martinez-Chantar, M.L.; Beraza, N. Inhibition of natural killer cells protects the liver against acute injury in the absence of glycine n-methyltransferase. Hepatology 2012, 56, 747–759. [Google Scholar]
- Zhao, M.; Chen, Y.H.; Dong, X.T.; Zhou, J.; Chen, X.; Wang, H.; Wu, S.X.; Xia, M.Z.; Zhang, C.; Xu, D.X. Folic acid protects against lipopolysaccharide-induced preterm delivery and intrauterine growth restriction through its anti-inflammatory effect in mice. PLoS ONE 2013, 8, e82713. [Google Scholar]
- Gao, L.; Siu, K.L.; Chalupsky, K.; Nguyen, A.; Chen, P.; Weintraub, N.L.; Galis, Z.; Cai, H. Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: Treatment with folic acid. Hypertension 2012, 59, 158–166. [Google Scholar]
- Hewitson, T.D. Renal tubulointerstitial fibrosis: Common but never simple. Am. J. Physiol. Ren. Physiol. 2009, 296, F1239–F1244. [Google Scholar]
- Pushpakumar, S.B.; Kundu, S.; Metreveli, N.; Sen, U. Folic acid mitigates angiotensin-ii-induced blood pressure and renal remodeling. PLoS ONE 2013, 8, e83813. [Google Scholar]
- Cao, L.; Lou, X.; Zou, Z.; Mou, N.; Wu, W.; Huang, X.; Tan, H. Folic acid attenuates hyperhomocysteinemia-induced glomerular damage in rats. Microvasc. Res. 2013, 89, 146–152. [Google Scholar] [PubMed]
- King, J.S.; Veltman, D.M.; Insall, R.H. The induction of autophagy by mechanical stress. Autophagy 2011, 7, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar]
- Martínez-López, N.; García-Rodríguez, J.L.; Varela-Rey, M.; Gutiérrez, V.; Fernández-Ramos, D.; Beraza, N.; Aransay, A.M.; Schlangen, K.; Lozano, J.J.; Aspichueta, P.; et al. Hepatoma cells from mice deficient in glycine n-methyltransferase have increased ras signaling and activation of liver kinase b1. Gastroenterology 2012, 143, 787–798.e13. [Google Scholar]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonchère, B.; Bélanger, A.; Guette, C.; Barré, B.; Coqueret, O. Stat3 as a new autophagy regulator. Jak-Stat 2013, 2, e24353. [Google Scholar] [PubMed] [Green Version]
- Yen, C.H.; Lu, Y.C.; Li, C.H.; Lee, C.M.; Chen, C.Y.; Cheng, M.Y.; Huang, S.F.; Chen, K.F.; Cheng, A.L.; Liao, L.Y.; et al. Functional characterization of glycine n-methyltransferase and its interactive protein depdc6/deptor in hepatocellular carcinoma. Mol. Med. 2012, 18, 286–296. [Google Scholar] [CrossRef]
- Sutter, B.M.; Wu, X.; Laxman, S.; Tu, B.P. Methionine inhibits autophagy and promotes growth by inducing the sam-responsive methylation of pp2a. Cell 2013, 154, 403–415. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0 | 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|---|
| Tubular injury | Normal | <10% | 10~25% | 25~50% | 50~75% | >75% |
| Leukocyte infiltration | Normal | <10% | 10~25% | 25~50% | 50~75% | >75% |
| Fibrotic area | Normal | <10% | 10~25% | 25~50% | 50~75% | >75% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, K.-L.; Chiang, C.-W.; Chen, Y.-M.A.; Yu, C.-C.; Lee, T.-S. Folic Acid Ameliorates Renal Injury in Experimental Obstructive Nephropathy: Role of Glycine N-Methyltransferase. Int. J. Mol. Sci. 2023, 24, 6859. https://doi.org/10.3390/ijms24076859
Kuo K-L, Chiang C-W, Chen Y-MA, Yu C-C, Lee T-S. Folic Acid Ameliorates Renal Injury in Experimental Obstructive Nephropathy: Role of Glycine N-Methyltransferase. International Journal of Molecular Sciences. 2023; 24(7):6859. https://doi.org/10.3390/ijms24076859
Chicago/Turabian StyleKuo, Ko-Lin, Chin-Wei Chiang, Yi-Ming Arthur Chen, Chih-Chin Yu, and Tzong-Shyuan Lee. 2023. "Folic Acid Ameliorates Renal Injury in Experimental Obstructive Nephropathy: Role of Glycine N-Methyltransferase" International Journal of Molecular Sciences 24, no. 7: 6859. https://doi.org/10.3390/ijms24076859