
Genetic Analysis of Multiple Primary Malignant Tumors in Women with Breast and Ovarian Cancer


Abstract
:1. Introduction
2. Results
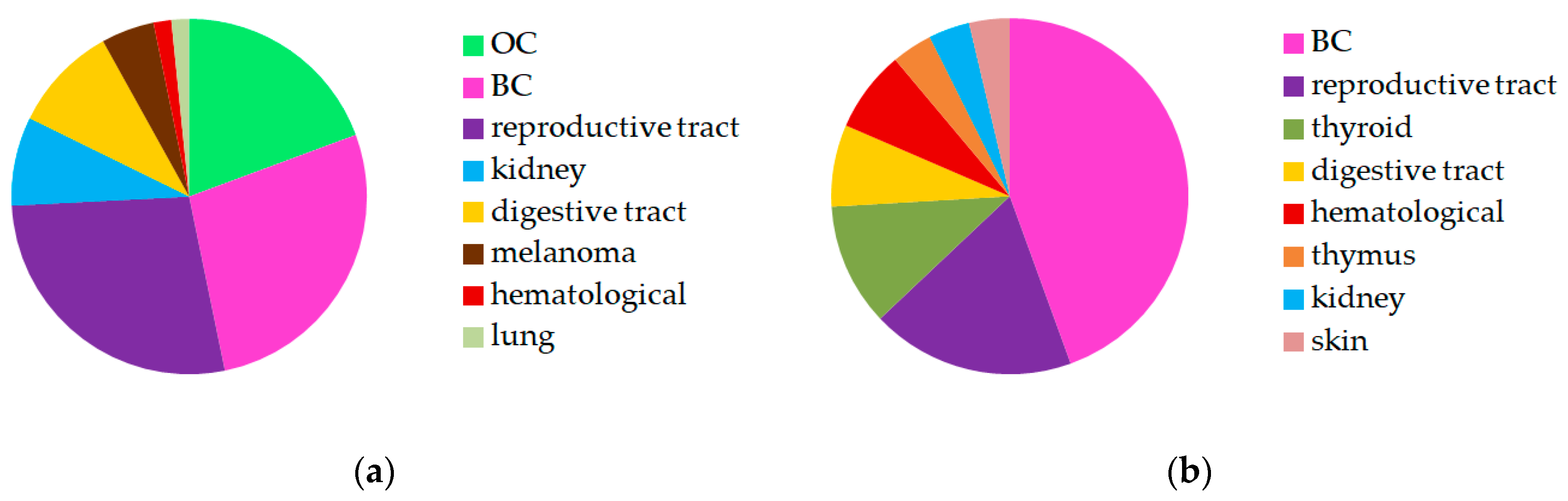
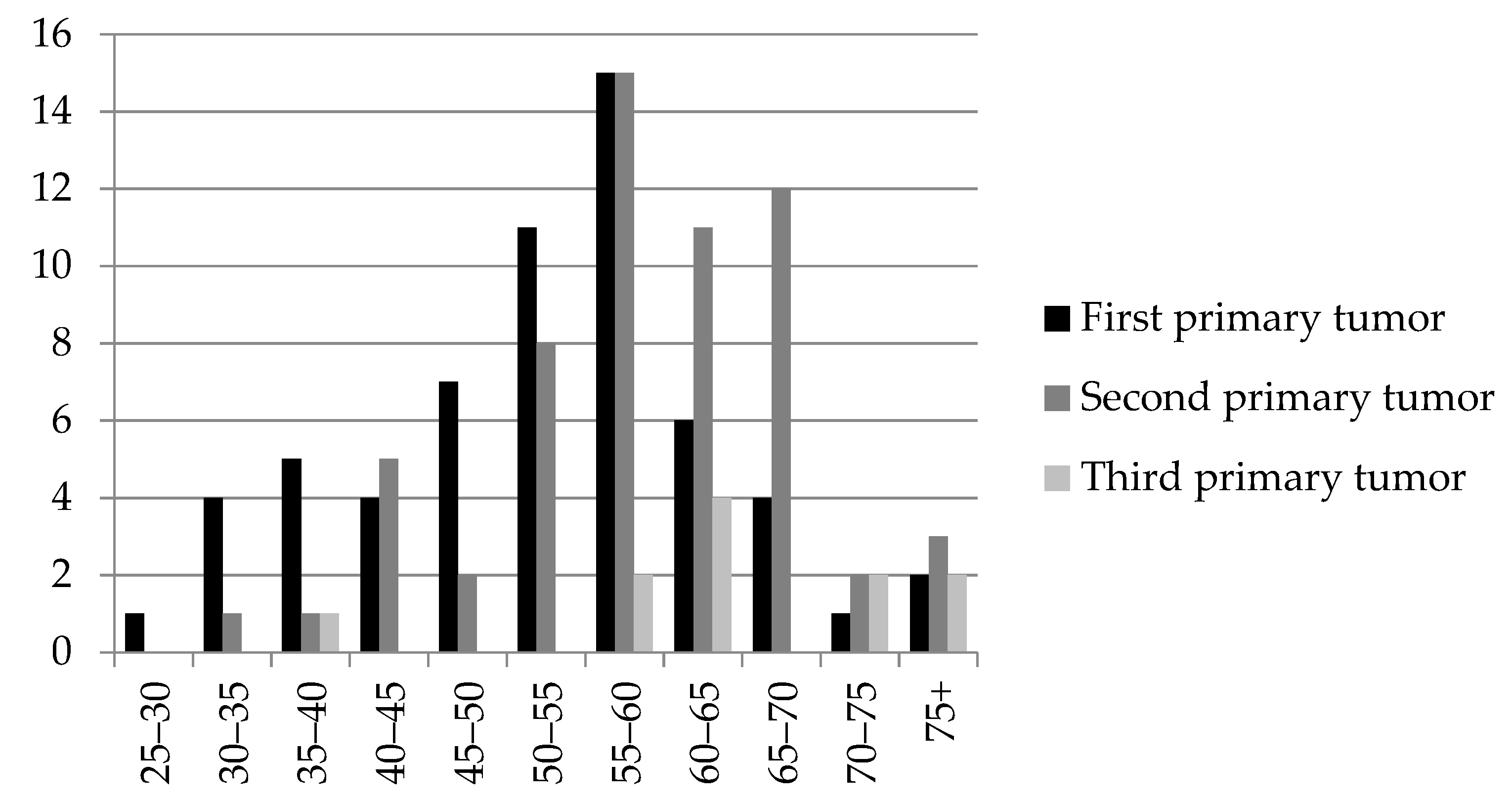
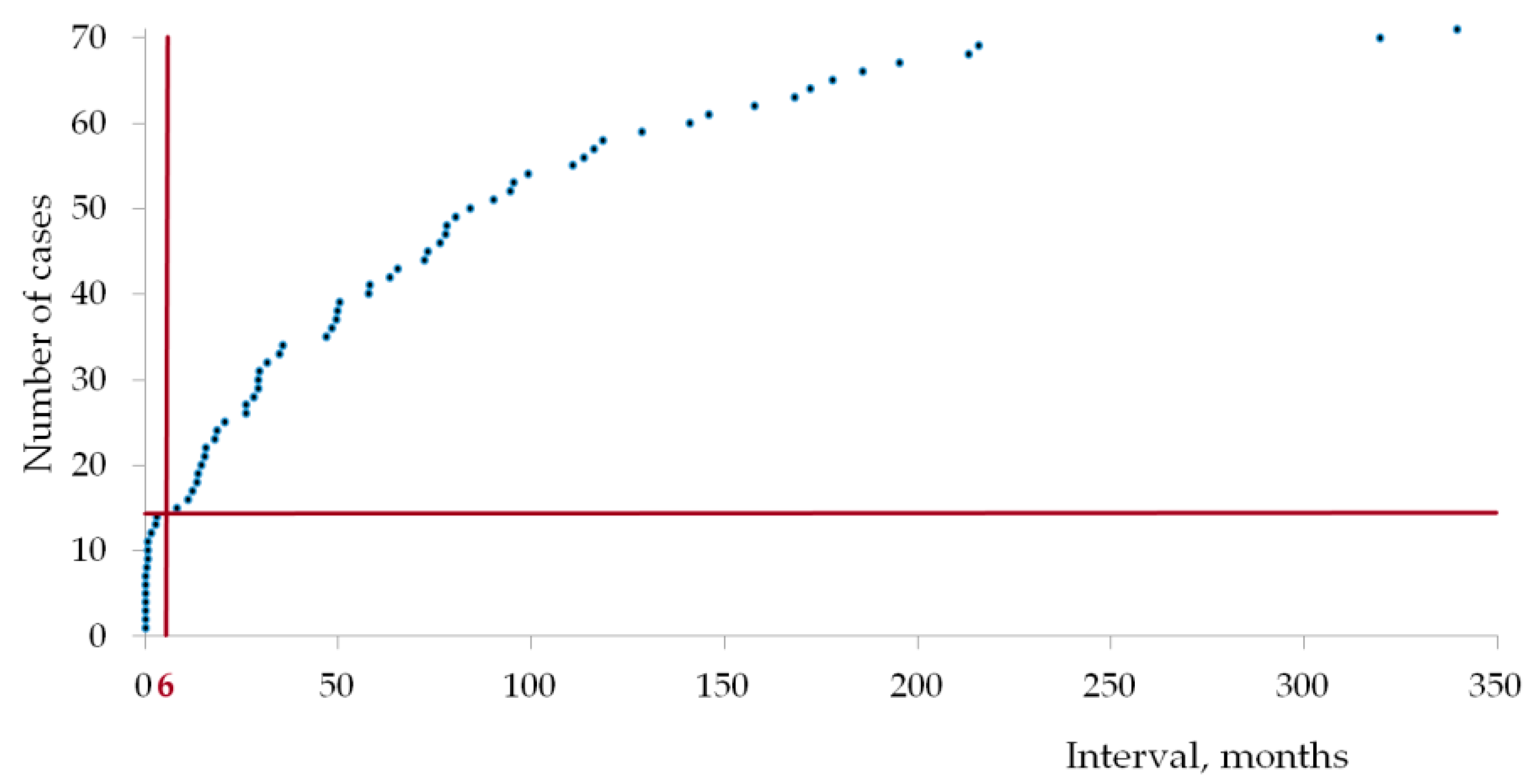
2.1. Patients’ Characteristics
2.2. Targeted Sequencing
3. Discussion
4. Materials and Methods
4.1. Patients and Their Histories
4.2. Morphological Examination
4.3. Targeted Sequencing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vogt, A.; Schmid, S.; Heinimann, K.; Frick, H.; Herrmann, C.; Cerny, T.; Omlin, A. Multiple primary tumours: Challenges and approaches, a review. ESMO Open 2017, 2, e000172. [Google Scholar] [CrossRef] [Green Version]
- Testori, A.; Cioffi, U.; De Simone, M.; Bini, F.; Vaghi, A.; Lemos, A.A.; Ciulla, M.M.; Alloisio, M. Multiple primary synchronous malignant tumors. BMC Res. Notes 2015, 8, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanjak, P.; Suktitipat, B.; Vorasan, N.; Juengwiwattanakitti, P.; Thiengtrong, B.; Songjang, C.; Therasakvichya, S.; Laiteerapong, S.; Chinswangwatanakul, V. Risks and cancer associations of metachronous and synchronous multiple primary cancers: A 25-year retrospective study. BMC Cancer 2021, 21, 1045. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.-Y.; Huang, C.-P.; Chen, W.-C. Synchronous/Metachronous Multiple Primary Malignancies: Review of Associated Risk Factors. Diagnostics 2022, 12, 1940. [Google Scholar] [CrossRef]
- Rice, J.M. Immunosuppression. In Tumour Site Concordance and Mechanisms of Carcinogenesis; Baan, R.A., Stewart, B.W., Straif, K., Eds.; International Agency for Research on Cancer: Lyon, France, 2019; pp. 159–162. [Google Scholar]
- Hong, H.; Wang, Q.; Li, J.; Liu, H.; Meng, X.; Zhang, H. Aging, Cancer and Immunity. J. Cancer 2019, 10, 3021–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcinelli, M.; Thaker, P.H.; Lutgendorf, S.K.; Conzen, S.D.; Flaherty, R.L.; Flint, M.S. The Role of Psychologic Stress in Cancer Initiation: Clinical Relevance and Potential Molecular Mechanisms. Cancer Res. 2021, 81, 5131–5140. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Mo, Y.; Wang, Y.; Xiang, B.; Liao, Q.; Zhou, M.; Li, X.; Li, Y.; Xiong, W.; Li, G.; et al. Chronic Stress Promotes Cancer Development. Front. Oncol. 2020, 10, 1492. [Google Scholar] [CrossRef]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, H.B.; Apffelstaedt, J.P. Counseling for male BRCA mutation carriers—A review. Breast 2008, 17, 441–450. [Google Scholar] [CrossRef]
- Gorodetska, I.; Serga, S.; Lahuta, T.; Ostapchenko, L.; Demydov, S.; Khranovska, N.; Skachkova, O.; Inomistova, M.; Kolesnik, O.; Svintsitsky, V.; et al. Prevalence of two BRCA1 mutations, 5382insC and 300T > G, in ovarian cancer patients from Ukraine. Fam. Cancer 2017, 16, 471–476. [Google Scholar] [CrossRef]
- Foretová, L.; Navrátilová, M.; Svoboda, M.; Vašíčková, P.; Hrabincová, E.S.; Házová, J.; Kleiblová, P.; Kleibl, Z.; Macháčková, E.; Palácová, M.; et al. Recommendations for Preventive Care for Women with Rare Genetic Cause of Breast and Ovarian Cancer. Klin. Onkol. 2019, 32, 6–13. [Google Scholar] [CrossRef]
- Cybulski, C.; Kluźniak, W.; Huzarski, T.; Wokołorczyk, D.; Kashyap, A.; Rusak, B.; Stempa, K.; Gronwald, J.; Szymiczek, A.; Bagherzadeh, M.; et al. The spectrum of mutations predisposing to familial breast cancer in Poland. Int. J. Cancer 2019, 145, 3311–3320. [Google Scholar] [CrossRef]
- Bogdanova, N.; Togo, A.V.; Ratajska, M.; Kluźniak, W.; Takhirova, Z.; Tarp, T.; Prokofyeva, D.; Bermisheva, M.; Yanus, G.A.; Gorodnova, T.V.; et al. Prevalence of the BLM nonsense mutation, p.Q548X, in ovarian cancer patients from Central and Eastern Europe. Fam. Cancer 2014, 14, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, A.P.; Iyevleva, A.G.; Preobrazhenskaya, E.V.; Mitiushkina, N.V.; Abysheva, S.N.; Suspitsin, E.N.; Kuligina, E.S.; Gorodnova, T.V.; Pfeifer, W.; Togo, A.V.; et al. High prevalence and breast cancer predisposing role of the BLM c.1642 C > T (Q548X) mutation in Russia. Int. J. Cancer 2011, 130, 2867–2873. [Google Scholar] [CrossRef]
- Wang, L.; Di, L.-J. BRCA1 and Estrogen/Estrogen Receptor in Breast Cancer: Where They Interact? Int. J. Biol. Sci. 2014, 10, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, J.L.; Nelson, A.C.; Lieberman, R.; Anderson, S.; Holt, J.T. Oestrogen-mediated phosphorylation and stabilization of BRCA2 protein in breast. J. Pathol. 2008, 217, 380–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulski, C.; Huzarski, T.; Byrski, T.; Gronwald, J.; Dębniak, T.; Jakubowska, A.; Górski, B.; Wokołorczyk, D.; Masojć, B.; Narod, S.; et al. Estrogen receptor status in CHEK2-positive breast cancers: Implications for chemoprevention. Clin. Genet. 2009, 75, 72–78. [Google Scholar] [CrossRef]
- Arora, A.; Abdel-Fatah, T.M.; Agarwal, D.; Doherty, R.; Moseley, P.M.; Aleskandarany, M.A.; Green, A.R.; Ball, G.; Alshareeda, A.T.; Rakha, E.A.; et al. Transcriptomic and Protein Expression Analysis Reveals Clinicopathological Significance of Bloom Syndrome Helicase (BLM) in Breast Cancer. Mol. Cancer Ther. 2015, 14, 1057–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Sokolenko, A.P.; Bogdanova, N.; Kluzniak, W.; Preobrazhenskaya, E.V.; Kuligina, E.S.; Iyevleva, A.G.; Aleksakhina, S.N.; Mitiushkina, N.V.; Gorodnova, T.V.; Bessonov, A.A.; et al. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Res. Treat. 2014, 145, 553–562. [Google Scholar] [CrossRef]
- Suspitsin, E.N.; Yanus, G.A.; Sokolenko, A.P.; Yatsuk, O.S.; Zaitseva, O.A.; Bessonov, A.A.; Ivantsov, A.O.; Heinstein, V.A.; Klimashevskiy, V.F.; Togo, A.V.; et al. Development of breast tumors in CHEK2, NBN/NBS1 and BLM mutation carriers does not commonly involve somatic inactivation of the wild-type allele. Med. Oncol. 2014, 31, 828. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Hajra, S.; Kaushik, A.; Rubahn, H.; Mishra, Y.; Kim, H. Smart nanomaterials as the foundation of a combination approach for efficient cancer theranostics. Mater. Today Chem. 2022, 26, 101182. [Google Scholar] [CrossRef]
- Homer, L.; Muller, M.; Dupré, P.-F.; Lucas, B.; Pradier, O. Uterine sarcoma associated with tamoxifen use after breast cancer: Review of the pathogenesis. J. Gynécologie Obs. Biol. La Reprod. 2009, 38, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Bagri, P.K.; Singh, D.; Singhal, M.K.; Singh, G.; Mathur, G.; Jakhar, S.L.; Beniwal, S.; Sharma, N.; Kumar, H.S.; Sharma, A.; et al. Double Primary Malignancies: A Clinical & Pathological Analysis Report from a Regional Cancer Institute in India. Iran. J. Cancer Prev. 2014, 7, 66–72. [Google Scholar]
- Xiao, L.; Cao, T.; Ou, J.; Liang, W. Clinical characteristics and prognostic analysis of multiple primary malignant neoplasms in female patients with breast cancer or genitalia malignancies. PeerJ 2022, 10, e13528. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of Breast and Ovarian Cancers among BRCA1 and BRCA2 Mutation Carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Biglia, N.; Sgandurra, P.; Bounous, V.E.; Maggiorotto, F.; Piva, E.; Pivetta, E.; Ponzone, R.; Pasini, B. Ovarian cancer in BRCA1 and BRCA2 gene mutation carriers: Analysis of prognostic factors and survival. Ecancermedicalscience 2016, 10, 639. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Shen, Y.; Zhang, Y.; Guo, F.; Li, Q.; Zhang, H.; Han, X.; Zhao, H.; Yang, Z. Metachronous Multiple Primary Carcinoma With Acute Promyelocytic Leukemia: 2 Cases Report and Literature Review. Front. Oncol. 2022, 12, 893319. [Google Scholar] [CrossRef]
- Zervoudakis, A.; Strickler, H.D.; Park, Y.; Xue, X.; Hollenbeck, A.; Schatzkin, A.; Gunter, M.J. Reproductive History and Risk of Colorectal Cancer in Postmenopausal Women. J. Natl. Cancer Inst. 2011, 103, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.-H.; Cotterchio, M.; Gallinger, S.; Knight, J.A.; Daftary, D. Family history of hormonal cancers and colorectal cancer risk: A case-control study conducted in Ontario. Int. J. Cancer 2009, 125, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Sundquist, J.; Sundquist, K.; Ji, J. Family history of breast cancer as a second primary malignancy in relatives: A nationwide cohort study. BMC Cancer 2021, 21, 1210. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Fallah, M.; Ji, J.; Sundquist, J.; Hemminki, K. The population impact of familial cancer, a major cause of cancer. Int. J. Cancer 2013, 134, 1899–1906. [Google Scholar] [CrossRef]
- Frank, C.; Fallah, M.; Sundquist, J.; Hemminki, A.; Hemminki, K. Population Landscape of Familial Cancer. Sci. Rep. 2015, 5, 12891. [Google Scholar] [CrossRef] [Green Version]
- Kemp, Z.; Turnbull, A.; Yost, S.; Seal, S.; Mahamdallie, S.; Poyastro-Pearson, E.; Warren-Perry, M.; Eccleston, A.; Tan, M.-M.; Teo, S.H.; et al. Evaluation of Cancer-Based Criteria for Use in Mainstream BRCA1 and BRCA2 Genetic Testing in Patients with Breast Cancer. JAMA Netw. Open 2019, 2, e194428. [Google Scholar] [CrossRef] [Green Version]
- Tennen, R.I.; Laskey, S.B.; Koelsch, B.L.; McIntyre, M.H.; Tung, J.Y. Identifying Ashkenazi Jewish BRCA1/2 founder variants in individuals who do not self-report Jewish ancestry. Sci. Rep. 2020, 10, 7669. [Google Scholar] [CrossRef] [PubMed]
- Laitman, Y.; Michaelson-Cohen, R.; Levi, E.; Chen-Shtoyerman, R.; Reish, O.; Josefsberg Ben-Yehoshua, S.; Bernstein-Molho, R.; Keinan-Boker, L.; Rosengarten, O.; Silverman, B.G.; et al. Uterine cancer in Jewish Israeli BRCA1/2 mutation carriers. Cancer 2018, 125, 698–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparri, M.L.; Taghavi, K.; Fiacco, E.; Zuber, V.; Di Micco, R.; Gazzetta, G.; Valentini, A.; Mueller, M.D.; Papadia, A.; Gentilini, O.D. Risk-Reducing Bilateral Salpingo-Oophorectomy for BRCA Mutation Carriers and Hormonal Replacement Therapy: If It Should Rain, Better a Drizzle than a Storm. Medicina 2019, 55, 415. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The First Tumor | The Second Tumor | The Third Tumor | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case | Histological Type | TNM Stage | Age | Histological Type | TNM Stage | Age | Histological Type | TNM Stage | Age | Family Cancer History: Localization and Age |
| Patients with BRCA1 5382insC | ||||||||||
| 1. | HGSOC 1 | T1a N0 M0 | 55 | IBC-NST 2 (grade II) | T1a N0 M0 | 56 | PMMNs: BC + OC (up to 50), E(M)C (52) | |||
| 2. | IBC-NST (grade III) | T1a N0 M0 | 32 | HGSOC | T3 N0 M0 | 58 | CRC 3 (70), BC (44), esophageal cancer (37) | |||
| 3. | IBC-NST (grade II; sin.) | T2 N0 M0 | 38 | IBC-NST (grade III; dex.) | T2 N0 M0 | 40 | BC (34), BC (50), bone sarcoma (48), lung cancer (31) | |||
| 4. | IBC-NST, (grade II) | T2 N0 M0 | 48 | LG 4 serous E(M)C 5 | T1b N0 M0 | 60 | BC (32), lung cancer (77) | |||
| 5. | IBC-NST (grade II; sin.) | T2 N0 M0 | 48 | IBC-NST (grade II; dex.) | T2 N0 M0 | 59 | HGSOC | T1b N0 M0 | 60 | stomach cancer (50) |
| 6. | IBC-NST (grade II; sin.) | T1 N0 M0 | 54 | IBC-NST (grade III; dex.) | T1 N1 M0 | 59 | HGSOC | T3c N0 M0 | 64 | BC (48), BC (60) |
| 7. | BC | T2 N1 M0 | 32 | IBC-NST (grade II) | T1a N0 M0 | 50 | Uterine leiomyosarcoma | T2 N0 M0 | 62 | Bilateral BC (41); thyroid cancer (52) |
| Patient with BRCA1 T300G | ||||||||||
| 8. | IBC-NST (grade II; sin.) | T2 N1 M0 | 59 | IBC-NST (grade II; dex.) | T2 N1 M0 | 63 | BC (60), stomach cancer (74), prostate cancer (52) | |||
| Patient with BRCA1 4153delA | ||||||||||
| 9. | IBC-NST (grade II) | T2 N1 M0 | 49 | HGSOC | T3 N0 M0 | 52 | CRC (78), cancer of unknown primary site (19), malignant brain tumor (51), nasopharyngeal carcinoma (48) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savkova, A.; Gulyaeva, L.; Gerasimov, A.; Krasil’nikov, S. Genetic Analysis of Multiple Primary Malignant Tumors in Women with Breast and Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 6705. https://doi.org/10.3390/ijms24076705
Savkova A, Gulyaeva L, Gerasimov A, Krasil’nikov S. Genetic Analysis of Multiple Primary Malignant Tumors in Women with Breast and Ovarian Cancer. International Journal of Molecular Sciences. 2023; 24(7):6705. https://doi.org/10.3390/ijms24076705
Chicago/Turabian StyleSavkova, Alina, Lyudmila Gulyaeva, Aleksey Gerasimov, and Sergey Krasil’nikov. 2023. "Genetic Analysis of Multiple Primary Malignant Tumors in Women with Breast and Ovarian Cancer" International Journal of Molecular Sciences 24, no. 7: 6705. https://doi.org/10.3390/ijms24076705