
Prognostic Value and Genome Signature of m6A/m5C Regulated Genes in Early-Stage Lung Adenocarcinoma

Abstract
:1. Introduction
2. Results
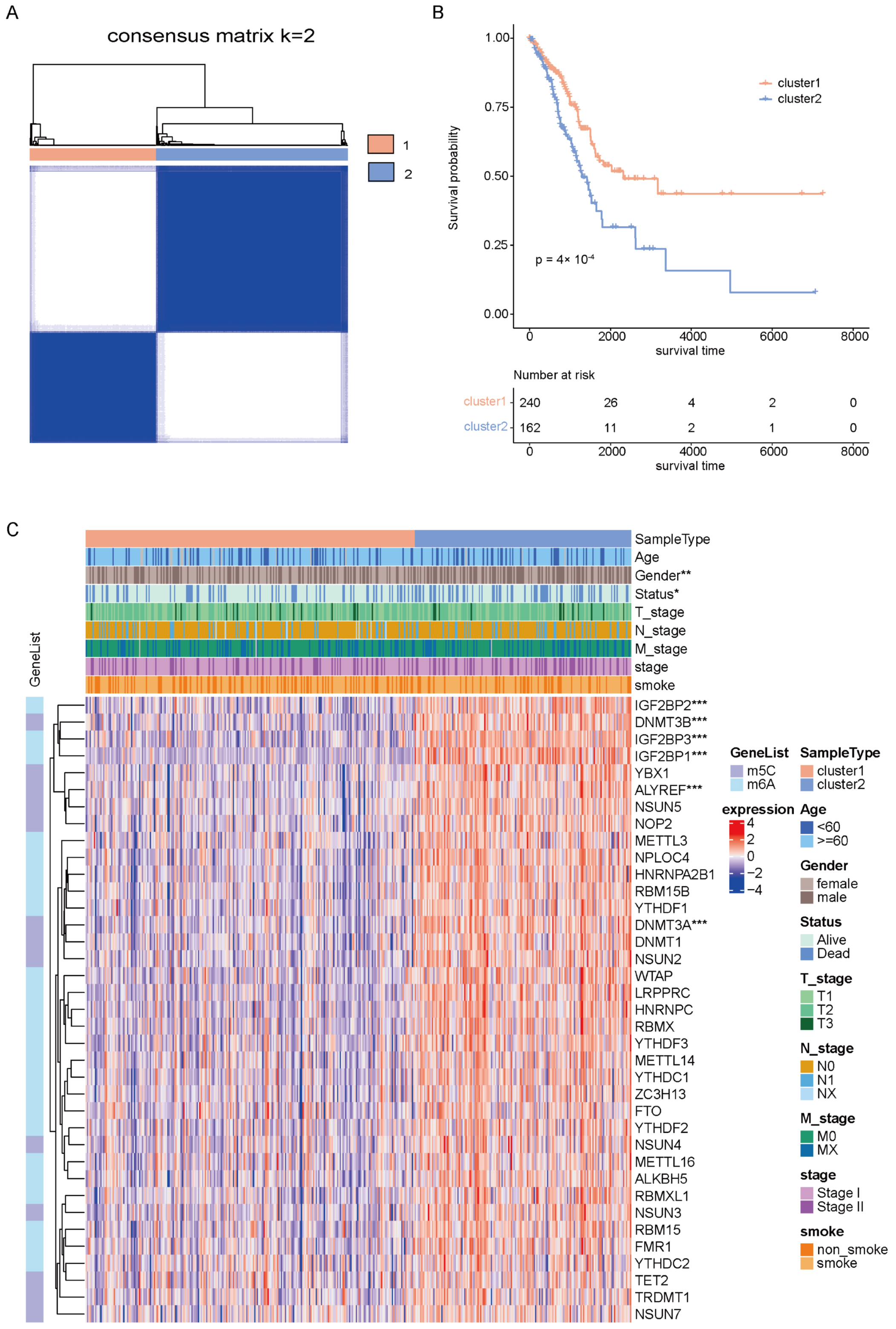
2.1. Identification of Molecular Clusters of Early-Stage LUAD Based on m6A/m5C-Related Genes
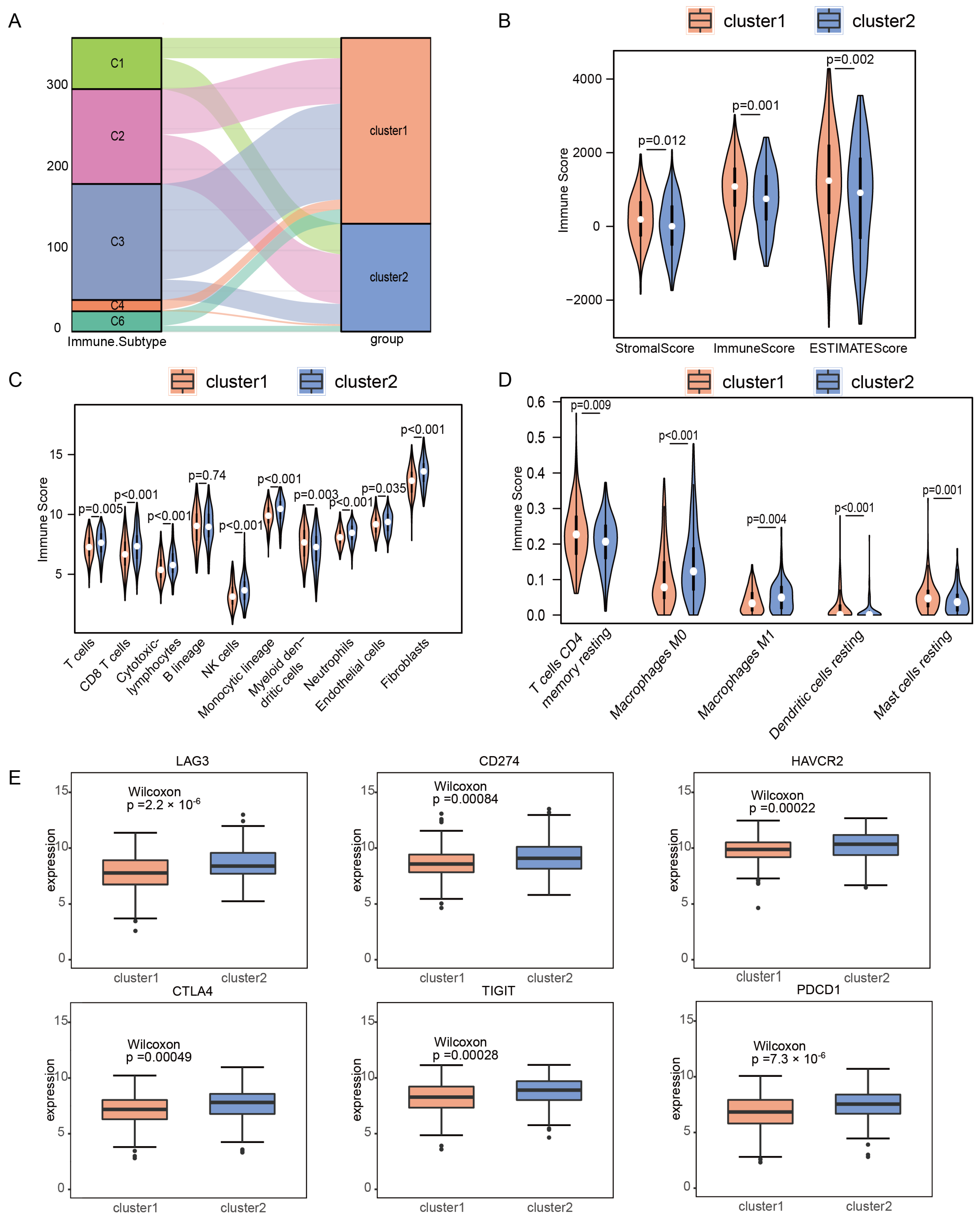
2.2. Immune Microenvironments of m6A/m5C Subtype in Early-Stage LUAD
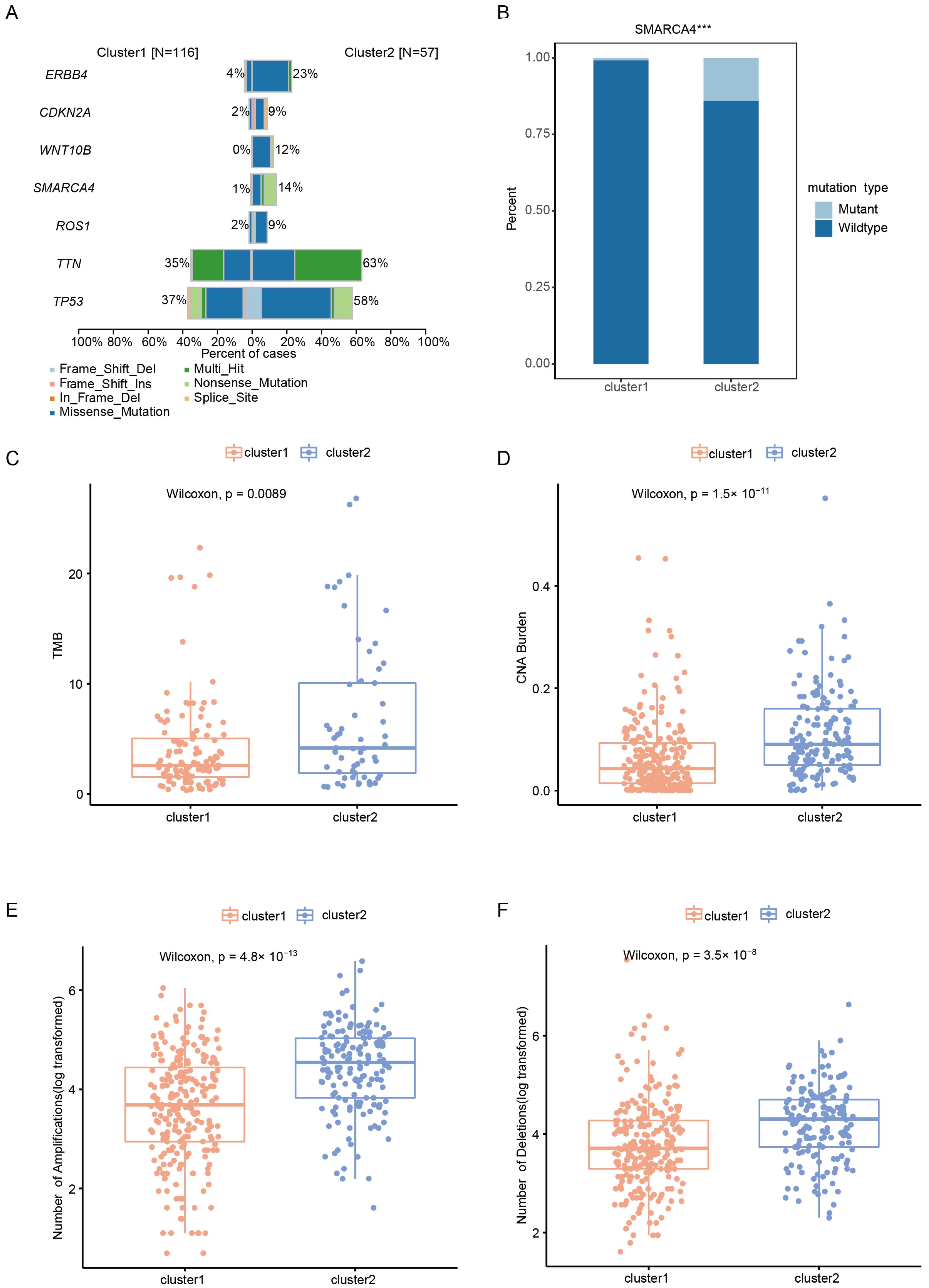
2.3. Somatic Alteration Landscape of Patients with Early-Stage LUAD in Different Clusters
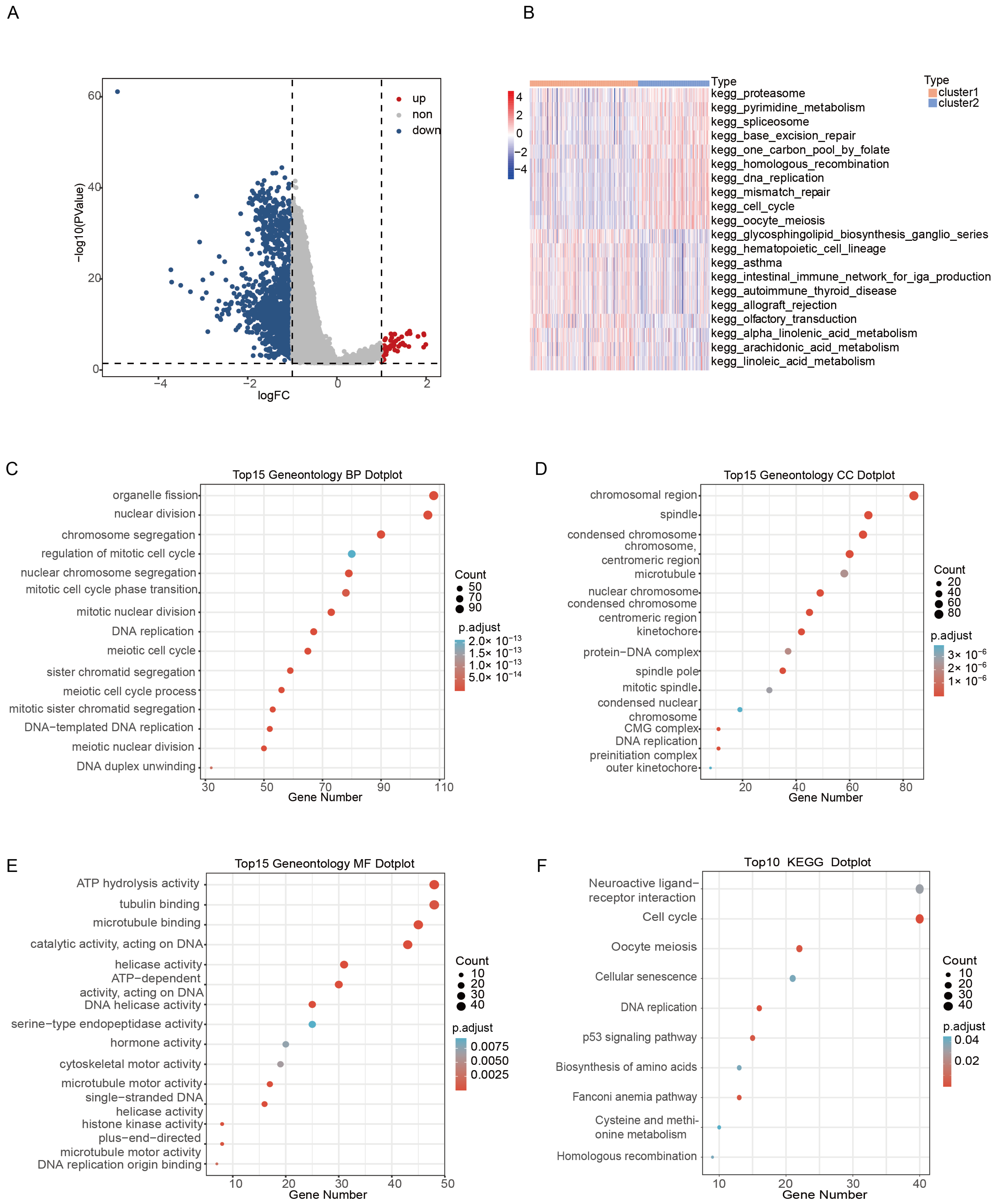
2.4. DEGs Identification and Functional Analysis between Clusters
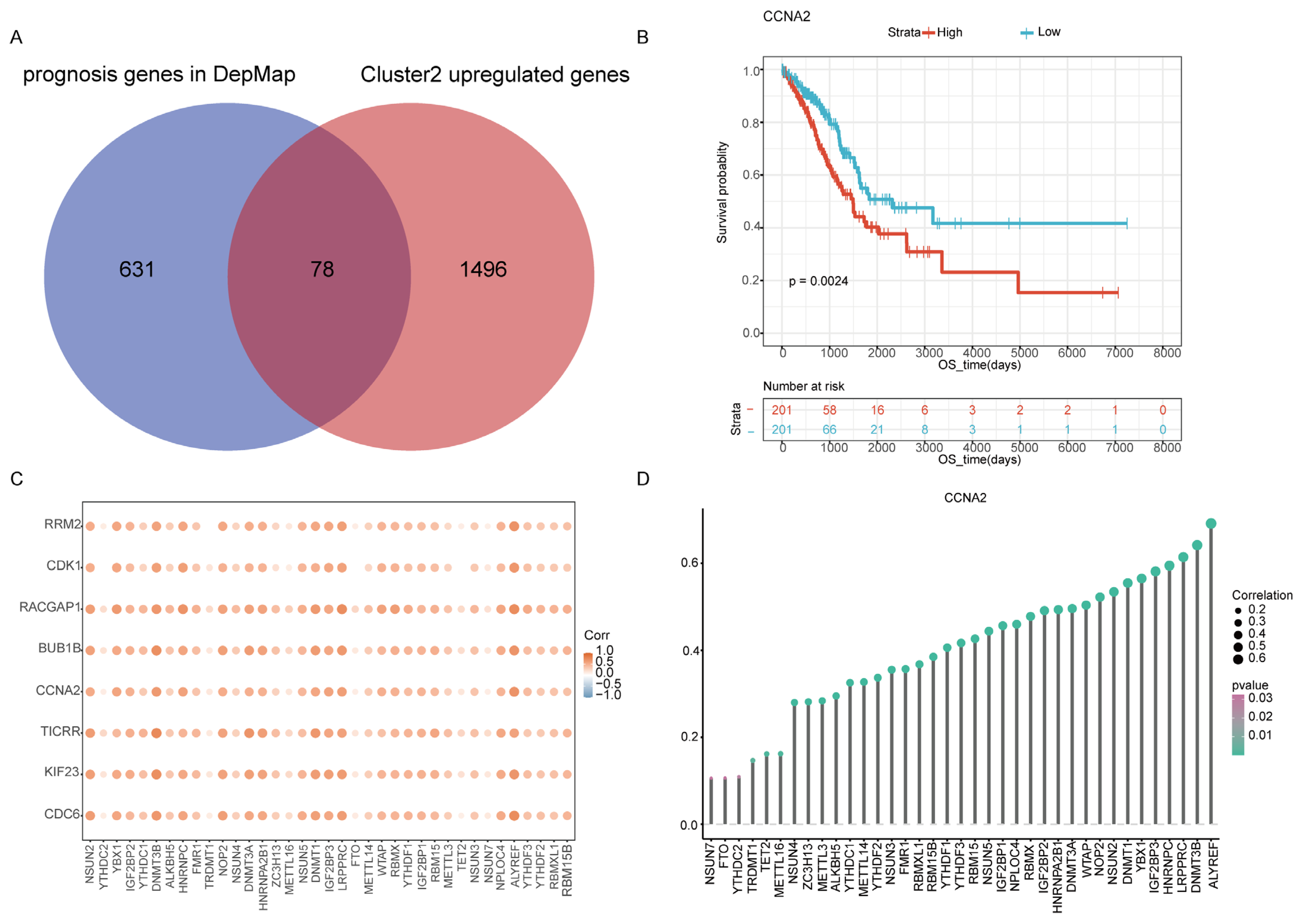
2.5. Identification Cluster2 Specific Hub Gene
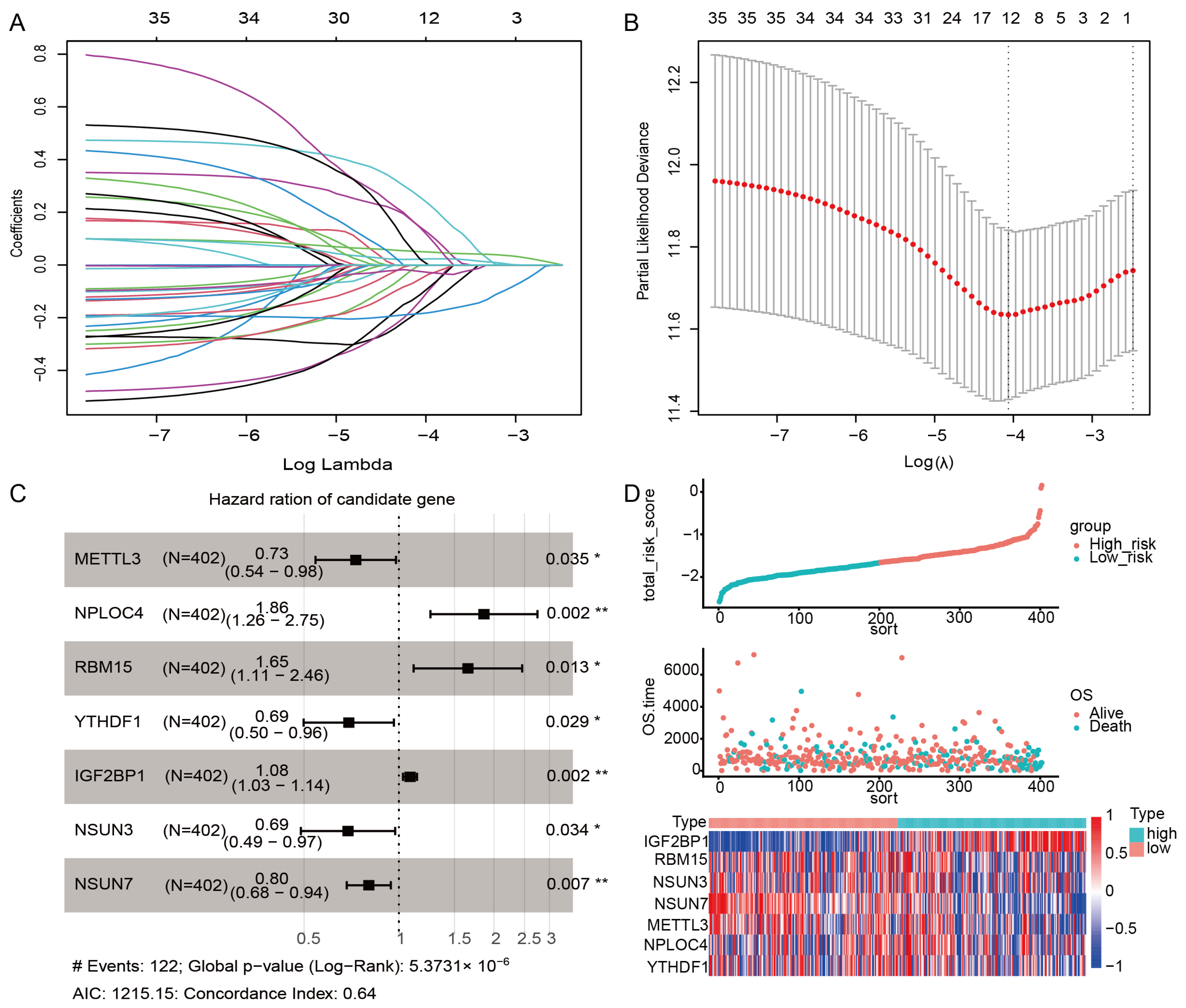
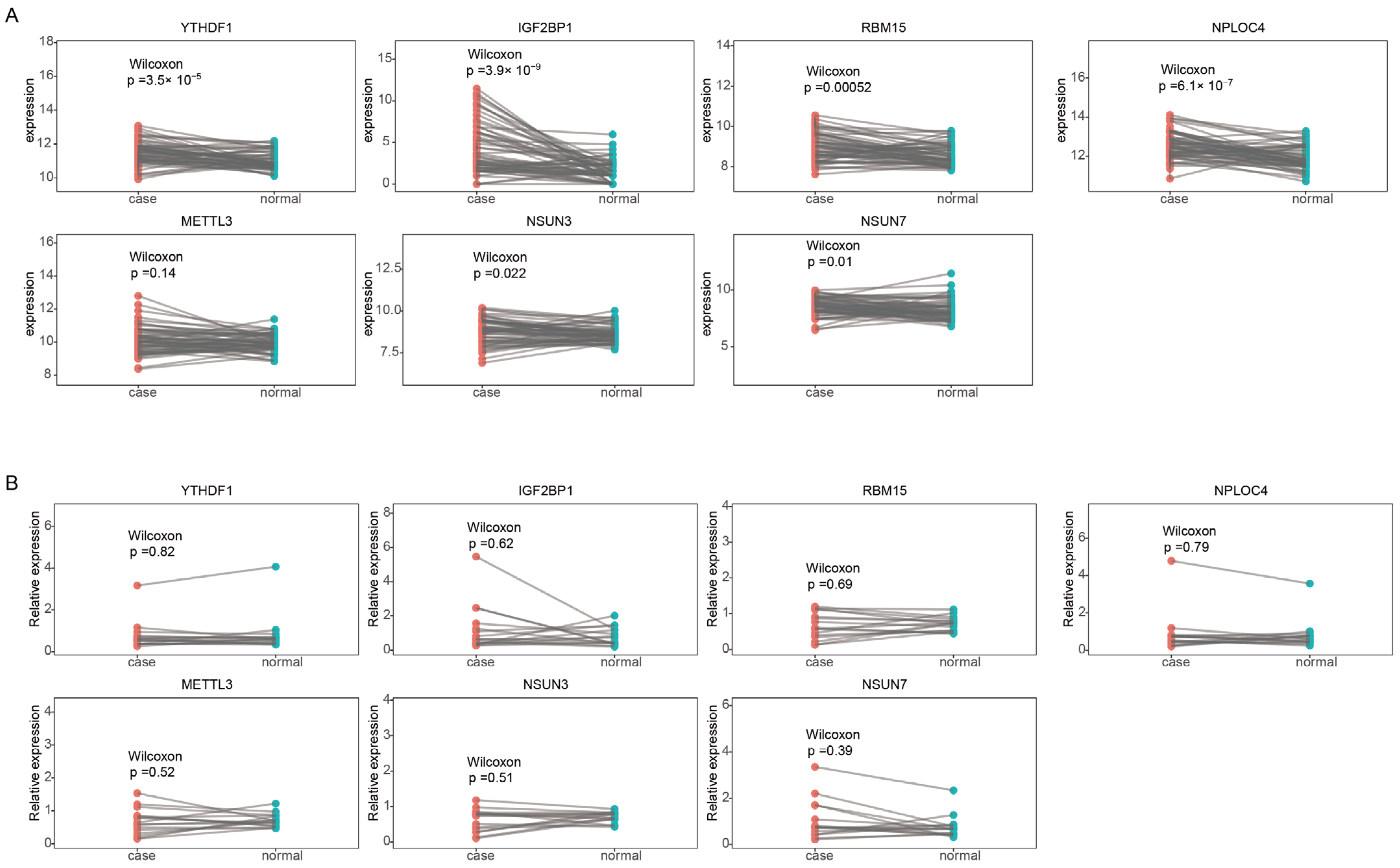
2.6. Construction of the m6A/m5C Related Prognostic Model
YTHDF1 + 0.074 × IGF2BP1 − 0.103 × NSUN3 − 0.216 × NSUN7.
3. Discussion
4. Materials and Methods
4.1. Publicly Available Cohort Datasets and Preprocessing
4.2. Consensus Clustering Analysis of m6A/m5C Regulated Gene
4.3. Correlation Analysis between Immune Infiltration Cells and m6A/m5C Subtypes
4.4. Somatic Mutation and Copy Number Alteration Analysis
4.5. Differentially Expressed Genes (DEGs), Enrichment Analysis, and Selection Hub Genes of m6A/m5C Subtypes
4.6. Construction of the m6A/m5C-Related Risk Score
4.7. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Nombela, P.; Miguel-López, B.; Blanco, S. The role of m6A, m5C and Ψ RNA modifications in cancer: Novel therapeutic opportunities. Mol. Cancer 2021, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Chen, J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Shen, Q.; Xu, Z.; Sun, G.; Wang, H.; Zhang, L. TFAP4 Activates IGF2BP1 and Promotes Progression of Non-Small Cell Lung Cancer by Stabilizing TK1 Expression through m6A Modification. Mol. Cancer Res. 2022, 20, 1763–1775. [Google Scholar] [CrossRef]
- Pan, J.; Huang, Z.; Xu, Y. m5C RNA Methylation Regulators Predict Prognosis and Regulate the Immune Microenvironment in Lung Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 657466. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Sun, C. M5C-Related lncRNA Predicts Lung Adenocarcinoma and Tumor Microenvironment Remodeling: Computational Biology and Basic Science. Front. Cell Dev. Biol. 2022, 10, 885568. [Google Scholar] [CrossRef] [PubMed]
- Chellamuthu, A.; Gray, S.G. The RNA Methyltransferase NSUN2 and Its Potential Roles in Cancer. Cells 2020, 9, 1758. [Google Scholar] [CrossRef]
- Lee, Y.; Choe, J.; Park, O.H.; Kim, Y.K. Molecular Mechanisms Driving mRNA Degradation by m6A Modification. Trends Genet 2020, 36, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A. The immune landscape of cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Liu, Y.; Wen, Y.; Zhou, C. Non-small cell lung cancer in China. Cancer Commun. 2022, 42, 937–970. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Yang, L.; Xiong, F.; He, Y.; Tang, Y.; Shi, L.; Fan, S.; Li, Z.; Zhang, S.; Gong, Z.; et al. Long non-coding RNA AFAP1-AS1 accelerates lung cancer cells migration and invasion by interacting with SNIP1 to upregulate c-Myc. Signal Transduct. Target. Ther. 2021, 6, 240. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Chu, Q.; Zheng, Q.; Jiang, S.; Bao, Z.; Su, Y.; Lu, J.; Li, L. Role of main RNA modifications in cancer: N6-methyladenosine, 5-methylcytosine, and pseudouridine. Signal Transduct. Target. Ther. 2022, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Wanna-Udom, S.; Terashima, M.; Lyu, H.; Ishimura, A.; Takino, T.; Sakari, M.; Tsukahara, T.; Suzuki, T. The m6A methyltransferase METTL3 contributes to Transforming Growth Factor-beta-induced epithelial-mesenchymal transition of lung cancer cells through the regulation of JUNB. Biochem. Biophys. Res. Commun. 2020, 524, 150–155. [Google Scholar] [CrossRef]
- Sheng, H.; Li, Z.; Su, S.; Sun, W.; Zhang, X.; Li, L.; Li, J.; Liu, S.; Lu, B.; Zhang, S.; et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis 2020, 41, 541–550. [Google Scholar] [CrossRef]
- Paggi, M.G.; Vona, R.; Abbruzzese, C.; Malorni, W. Gender-related disparities in non-small cell lung cancer. Cancer Lett. 2010, 298, 1–8. [Google Scholar] [CrossRef]
- Yang, M.; Lin, C.; Wang, Y.; Chen, K.; Zhang, H.; Li, W. Identification of a cytokine-dominated immunosuppressive class in squamous cell lung carcinoma with implications for immunotherapy resistance. Genome Med. 2022, 14, 72. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Belk, J.A.; Daniel, B.; Satpathy, A.T. Epigenetic regulation of T cell exhaustion. Nat. Immunol. 2022, 23, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; Ning, J.; Liu, W.; Li, K.; Qian, B.; Xu, D.; Wu, Y.; Zhang, D.; Cui, W. YTHDF1 Promotes Cyclin B1 Translation through m6A Modulation and Contributes to the Poor Prognosis of Lung Adenocarcinoma with KRAS/TP53 Co-Mutation. Cells 2021, 10, 1669. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Liu, Y.; Yao, Y.; Zhao, Y.; Bi, Z.; Jiang, Q.; Liu, Q.; Cai, M.; Wang, F.; Wang, Y.; et al. FTO regulates adipogenesis by controlling cell cycle progression via m6A-YTHDF2 dependent mechanism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1323–1330. [Google Scholar] [CrossRef]
- Yang, J.; Qian, X.; Qiu, Q.; Xu, L.; Pan, M.; Li, J.; Ren, J.; Lu, B.; Qiu, T.; Chen, E.; et al. LCAT1 is an oncogenic LncRNA by stabilizing the IGF2BP2-CDC6 axis. Cell Death Dis. 2022, 13, 877. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Li, Y.; Ming, R.; Wei, J.; Du, P.; Zhou, P.; Zong, S.; Xiao, H. The Prognostic Value and Immune Landscapes of a m6A/m5C/m1A-Related LncRNAs Signature in Head and Neck Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 718974. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Matteson, R. DepMap: Dependency Mapping of Applications Using Operating System Events. Master’s Thesis, California Polytechnic State University, San Luis Obispo, CA, USA, 2010. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R.; Narasimhan, B.; Tay, K.; Simon, N.; Qian, J. Package ‘Glmnet’. CRAN R Repositary 2021. Available online: https://cran.r-project.org/web/packages/glmnet/index.html (accessed on 1 September 2022).
- Ranstam, J.; Cook, J. LASSO regression. J. Br. Surg. 2018, 105, 1348. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Lorent, M.; Giral, M.; Foucher, Y. Net time-dependent ROC curves: A solution for evaluating the accuracy of a marker to predict disease-related mortality. Stat. Med. 2014, 33, 2379–2389. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M. A Package for Survival Analysis in R. 2019. Available online: https://CRAN.R-project.org/package=survival (accessed on 18 August 2022).
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves Using ‘ggplot2′. R Package Version 0.3.1. The Comprehensive R Archive Network. 2017. Available online: https://CRAN.R-project.org/package=survminer (accessed on 13 August 2022).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequences | |
|---|---|---|
| METTL3 | Forward | ACAGGCCGTACAGGTCAC |
| Reverse | ATCACATCACAATCCAGACCCT | |
| NPLOC4 | Forward | CCTCGTCTCAGAAGATACCCGAA |
| Reverse | TTGGGATGCTTGTTCTGGAAGTC | |
| NSUN3 | Forward | TGCCCAGCCTGAAATGTTTG |
| Reverse | TGAGAGTCAGAAGAAAACAACCAG | |
| NSUN7 | Forward | GCAGCATTGGCAAGATGTCGA |
| Reverse | TGGAGGCCCTTAGTTCCTGT | |
| RBM15 | Forward | GGCTGCCTGAGGAGAGTGGAG |
| Reverse | CGGCTACTGCTCAATTCTGGACTG | |
| YTHDF1 | Forward | CCCAGAGAACAAAAGGACAAGA |
| Reverse | TGTCCAGTAAGGTAGGGCTCAA | |
| IGF2BP1 | Forward | ATGGTTATCATCACTGGACCG |
| Reverse | TGTGGGTCTCCAGCTTCACT | |
| GAPDH | Forward | TGCACCACCAACTGCTTAGC |
| Reverse | GGCATGGACTGTGGTCATGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, L.; Wang, Y.; Tian, J.; Song, W.; Li, L.; Che, G. Prognostic Value and Genome Signature of m6A/m5C Regulated Genes in Early-Stage Lung Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 6520. https://doi.org/10.3390/ijms24076520
Tian L, Wang Y, Tian J, Song W, Li L, Che G. Prognostic Value and Genome Signature of m6A/m5C Regulated Genes in Early-Stage Lung Adenocarcinoma. International Journal of Molecular Sciences. 2023; 24(7):6520. https://doi.org/10.3390/ijms24076520
Chicago/Turabian StyleTian, Long, Yan Wang, Jie Tian, Wenpeng Song, Lu Li, and Guowei Che. 2023. "Prognostic Value and Genome Signature of m6A/m5C Regulated Genes in Early-Stage Lung Adenocarcinoma" International Journal of Molecular Sciences 24, no. 7: 6520. https://doi.org/10.3390/ijms24076520