
The Role of Cytokines in Cholesterol Accumulation in Cells and Atherosclerosis Progression
 , , , ,
, , , ,
Abstract
:1. Introduction
2. What Are Cytokines?
2.1. Interferons
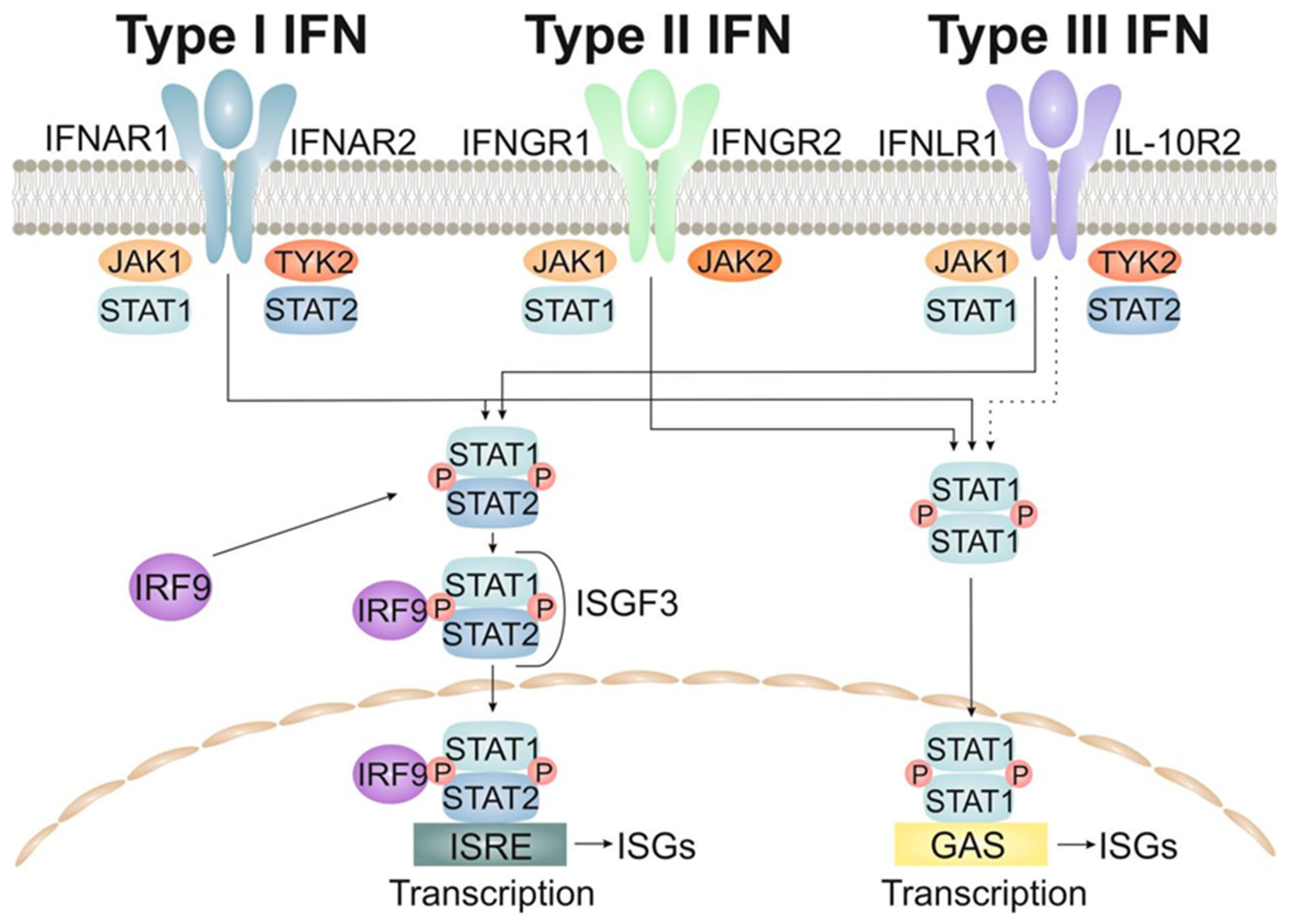
2.1.1. The Type-I Interferons (IFNs)
2.1.2. The Type-II Interferons
2.1.3. The Type-III Interferons
2.2. The Interleukins
2.3. The Tumor Necrosis Factor Superfamily
2.4. Chemokines
3. How Cytokines Impact Cholesterol Metabolism
3.1. Impact of Interferons on Cholesterol Metabolism
3.2. Impact of Interleukins on Cholesterol Metabolism
3.2.1. The Interleukin-1 (IL-1) Cytokine Family
3.2.2. Interleukin 4
3.2.3. Interleukin 5
3.2.4. Interleukin 6
3.2.5. Interleukin 7
3.2.6. Interleukin 8
3.2.7. Interleukin 10
3.2.8. Interleukin 12
3.2.9. Interleukin 13
3.2.10. Interleukin 15
3.2.11. Interleukin 17
3.2.12. Interleukin 22
3.3. Impact of the Tumor Necrosis Factor Superfamily (TNFSF) on Cholesterol Metabolism
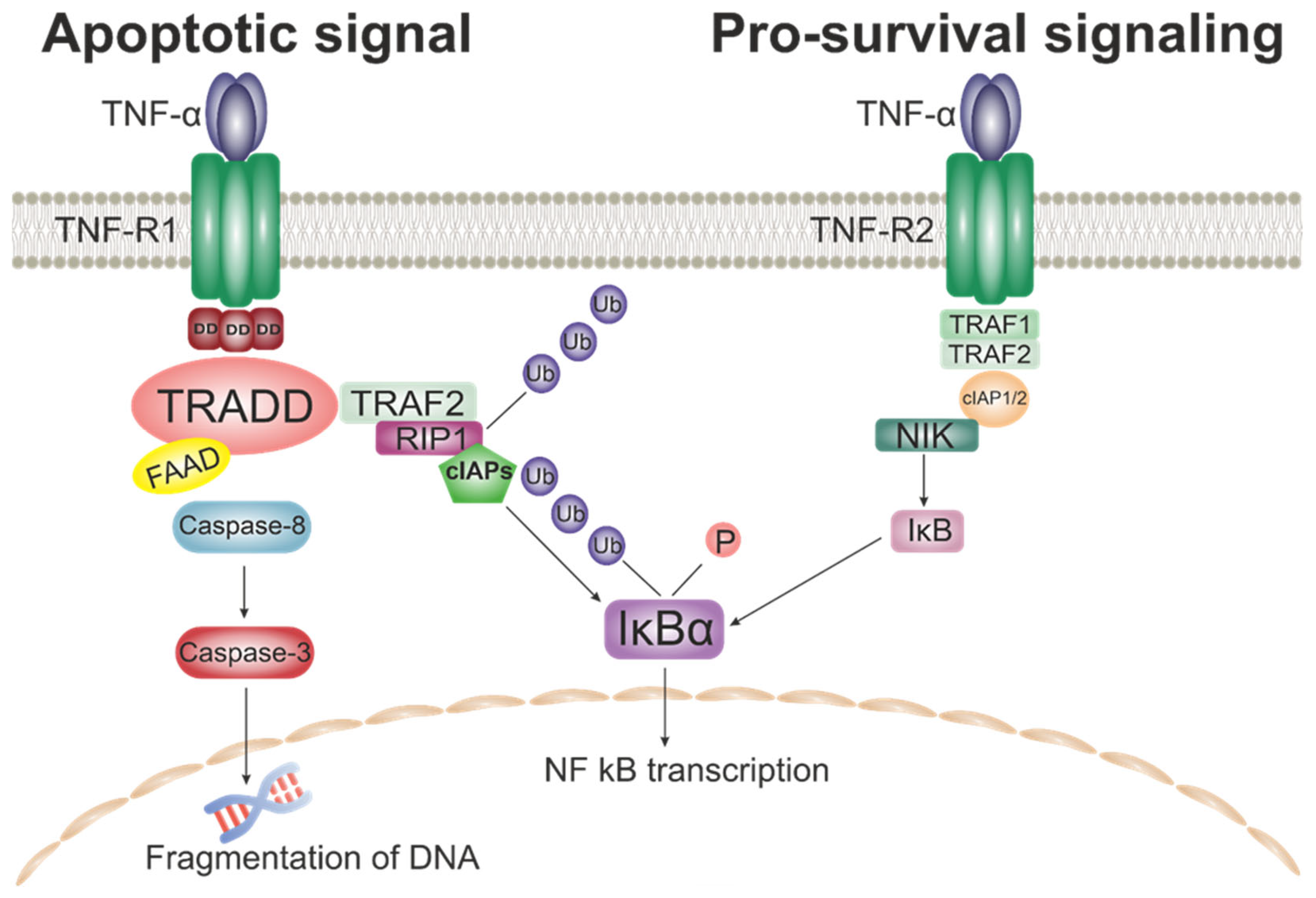
3.3.1. TNF-α
3.3.2. TRAIL
3.4. Impact of Chemokines on Cholesterol Metabolism
3.5. Impact of Colony-Stimulating Factors (CSF) on Cholesterol Metabolism
3.6. Impact of Transforming Growth Factors (TGF) on Cholesterol Metabolism
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 11 February 2023).
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and Senolytics in Cardiovascular Disease: Promise and Potential Pitfalls. Mech Ageing Dev. 2021, 198, 111540. [Google Scholar] [CrossRef]
- Li, J.; Han, Y.; Jing, J.; Tu, S.; Chen, W.; Reiber, J.H.C.; Chen, Y. Non-Culprit Coronary Lesions in Young Patients Have Higher Rates of Atherosclerotic Progression. Int. J. Cardiovasc. Imaging 2015, 31, 889–897. [Google Scholar] [CrossRef]
- Singh, S.S.; Pilkerton, C.S.; Shrader, C.D.; Frisbee, S.J. Subclinical Atherosclerosis, Cardiovascular Health, and Disease Risk: Is There a Case for the Cardiovascular Health Index in the Primary Prevention Population? BMC Public Health 2018, 18, 429. [Google Scholar] [CrossRef]
- Csige, I.; Ujvárosy, D.; Szabó, Z.; Lorincz, I.; Paragh, G.; Harangi, M.; Somodi, S.; Santulli, G. The Impact of Obesity on the Cardiovascular System. J. Diabetes Res. 2018, 2018, 3407306. [Google Scholar] [CrossRef] [Green Version]
- Maas, A.H.E.M.; Appelman, Y.E.A. Gender Differences in Coronary Heart Disease. Neth. Heart J. 2010, 18, 598–603. [Google Scholar] [CrossRef]
- Rubin, J.B.; Borden, W.B. Coronary Heart Disease in Young Adults. Curr. Atheroscler. Rep. 2012, 14, 140–149. [Google Scholar] [CrossRef]
- He, J.; Zhu, Z.; Bundy, J.D.; Dorans, K.S.; Chen, J.; Hamm, L.L. Trends in Cardiovascular Risk Factors in US Adults by Race and Ethnicity and Socioeconomic Status, 1999–2018. JAMA 2021, 326, 1286–1298. [Google Scholar] [CrossRef]
- Markin, A.M.; Sobenin, I.A.; Grechko, A.V.; Zhang, D.; Orekhov, A.N. Cellular Mechanisms of Human Atherogenesis: Focus on Chronification of Inflammation and Mitochondrial Mutations. Front. Pharm. 2020, 11, 642. [Google Scholar] [CrossRef]
- Horio, E.; Kadomatsu, T.; Miyata, K.; Arai, Y.; Hosokawa, K.; Doi, Y.; Ninomiya, T.; Horiguchi, H.; Endo, M.; Tabata, M.; et al. Role of Endothelial Cell-Derived Angptl2 in Vascular Inflammation Leading to Endothelial Dysfunction and Atherosclerosis Progression. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2020, 11, 613780. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Ivanova, E.A.; Markin, A.M.; Nikiforov, N.G.; Sobenin, I.A. Genetics of Arterial-Wall-Specific Mechanisms in Atherosclerosis: Focus on Mitochondrial Mutations. Curr. Atheroscler. Rep. 2020, 22, 54. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Brinck-Jensen, N.S.; Zang, M.; Chen, K. Monocyte-Derived Dendritic Cells: Targets as Potent Antigen-Presenting Cells for the Design of Vaccines against Infectious Diseases. Int. J. Infect. Dis. 2014, 19, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Zhang, W.; Zhao, Y.; Wang, F.; Liu, S.; Liu, L.; Zhao, L.; Lu, W.; Li, M.; Xu, Y. Dendritic Cells and T Cells, Partners in Atherogenesis and the Translating Road Ahead. Front. Immunol. 2020, 11, 1456. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Coornaert, I.; Puylaert, P.; de Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 Coordinates NLRP3 Inflammasome Activation by Facilitating Intracellular Nucleation of Soluble Ligands into Particulate Ligands in Sterile Inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nũez, G.; Schnurr, M.; et al. NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Strittmatter, G.E.; Garstkiewicz, M.; Sand, J.; Beer, H.D. Caspase-1: The Inflammasome and Beyond. Innate Immun. 2014, 20, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.S. Caspase-11 Non-Canonical Inflammasome: Emerging Activator and Regulator of Infection-Mediated Inflammatory Responses. Int. J. Mol. Sci. 2020, 21, 2736. [Google Scholar] [CrossRef] [Green Version]
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 Is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome Activation and Regulation: Toward a Better Understanding of Complex Mechanisms. Cell. Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.W.E.; Ramji, D.P. Cytokines: Roles in Atherosclerosis Disease Progression and Potential Therapeutic Targets. Future Med. Chem. 2016, 8, 1317–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C. Cytokine Regulation and Function in T Cells. Annu. Rev. Immunol. 2021, 39, 51–76. [Google Scholar] [CrossRef]
- Ożańska, A.; Szymczak, D.; Rybka, J. Pattern of Human Monocyte Subpopulations in Health and Disease. Scand. J. Immunol. 2020, 92, e12883. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Thon, J.N.; Italiano, J.E. Platelets: Production, Morphology and Ultrastructure. Handb. Exp. Pharm. 2012, 210, 3–22. [Google Scholar] [CrossRef]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. [Google Scholar] [CrossRef]
- Gomez, D.; Owens, G.K. Smooth Muscle Cell Phenotypic Switching in Atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, M.C.; Berry, R.; Holtrup, B.; Sebo, Z.; Nelson, T.; Fretz, J.A.; Lindskog, D.; Kaplan, J.L.; Ables, G.; Rodeheffer, M.S.; et al. Bone Marrow Adipocytes. Adipocyte 2017, 6, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Fatkhullina, A.R.; Peshkova, I.O.; Koltsova, E.K. The Role of Cytokines in the Development of Atherosclerosis. Biochemistry 2016, 81, 1358–1370. [Google Scholar] [CrossRef] [Green Version]
- Gale, M., Jr. Action of Mustards on Interfering, Hemagglutinating and Eluting Capacities of Innuenza a (PR8) Virus. J. Immunol. 2015, 195, 1909–1910. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.T.; Gale, M. SnapShot: Interferon Signaling. Cell 2015, 163, 1808–1808.e1. [Google Scholar] [CrossRef]
- Bolko, L.; Jiang, W.; Tawara, N.; Landon-Cardinal, O.; Anquetil, C.; Benveniste, O.; Allenbach, Y. The Role of Interferons Type I, II and III in Myositis: A Review. Brain Pathol. 2021, 31, e12955. [Google Scholar] [CrossRef]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III Interferons Disrupt Lung Epithelial Repair during Recovery from Viral Infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef]
- Broggi, A.; Granucci, F.; Zanoni, I. Type III Interferons: Balancing Tissue Tolerance and Resistance to Pathogen Invasion. J. Exp. Med. 2020, 217, e20190295. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; PLoSs, A. Decoding Type I and III Interferon Signalling during Viral Infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, Interferon-like Cytokines, and Their Receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Chen, H.J.; Tas, S.W.; de Winther, M.P.J. Type-I Interferons in Atherosclerosis. J. Exp. Med. 2020, 217, e20190459. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.; Steinhoff, U.; Reis, L.F.L.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional Role of Type I and Type II Interferons in Antiviral Defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef] [PubMed]
- Crisler, W.J.; Lenz, L.L. Crosstalk between Type I and II Interferons in Regulation of Myeloid Cell Responses during Bacterial Infection. Curr. Opin. Immunol. 2018, 54, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Taniguchi, T.; Yanai, H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb. Perspect. Biol. 2018, 10, a028423. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-Lambda (IFN-l) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PLoS Pathog. 2008, 4, 1000017. [Google Scholar] [CrossRef]
- Odendall, C.; Kagan, J.C. The Unique Regulation and Functions of Type III Interferons in Antiviral Immunity. Curr. Opin. Virol. 2015, 12, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Manivasagam, S.; Klein, R.S. Type III Interferons: Emerging Roles in Autoimmunity. Front. Immunol. 2021, 12, 764062. [Google Scholar] [CrossRef]
- Sims, J.E.; Smith, D.E. The IL-1 Family: Regulators of Immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Flusberg, D.A.; Sorger, P.K. Surviving Apoptosis: Life-Death Signaling in Single Cells. Trends Cell Biol. 2015, 25, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Nash, M.; McGrath, J.P.; Cartland, S.P.; Patel, S.; Kavurma, M.M. Tumour Necrosis Factor Superfamily Members in Ischaemic Vascular Diseases. Cardiovasc. Res. 2019, 115, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Ferrao, R.; Wu, H. Helical Assembly in the Death Domain (DD) Superfamily. Curr. Opin. Struct. Biol. 2012, 22, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Vetreno, R.P.; Crews, F.T. Hippocampal TNF-Death Receptors, Caspase Cell Death Cascades, and IL-8 in Alcohol Use Disorder. Mol. Psychiatry 2021, 26, 2254–2262. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.L.; Hughes, M.A.; Meng, X.; Sarnowska, N.A.; Powley, I.R.; Jukes-Jones, R.; Dinsdale, D.; Ragan, T.J.; Fairall, L.; Schwabe, J.W.R.; et al. Cryo-EM Structural Analysis of FADD: Caspase-8 Complexes Defines the Catalytic Dimer Architecture for Co-Ordinated Control of Cell Fate. Nat. Commun. 2021, 12, 819. [Google Scholar] [CrossRef]
- Park, Y.H.; Han, C.W.; Jeong, M.S.; Jang, S.B. DED Interaction of FADD and Caspase-8 in the Induction of Apoptotic Cell Death. J. Microbiol. Biotechnol. 2022, 32, 1034. [Google Scholar] [CrossRef]
- Mnich, K.; Koryga, I.; Pakos-Zebrucka, K.; Thomas, M.; Logue, S.E.; Eriksson, L.A.; Gorman, A.M.; Samali, A. The Stressosome, a Caspase-8-activating Signalling Complex Assembled in Response to Cell Stress in an ATG5-mediated Manner. J. Cell. Mol. Med. 2021, 25, 8809. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Sun, S.C. The Non-Canonical NF-ΚB Pathway in Immunity and Inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Kusters, P.J.H.; Lutgens, E. Cytokines and Immune Responses in Murine Atherosclerosis. Methods Mol. Biol. 2015, 1339, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Ware, C.F.; Croft, M.; Neil, G.A. Realigning the LIGHT Signaling Network to Control Dysregulated Inflammation. J. Exp. Med. 2022, 219, e20220236. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, X.; Zhu, G.; Liu, H.; Chen, J.; Wang, Y.; He, X. Quercetin Inhibits TNF-α Induced HUVECs Apoptosis and Inflammation via Downregulating NF-KB and AP-1 Signaling Pathway in vitro. Medicine 2020, 99, e22241. [Google Scholar] [CrossRef]
- Willemsen, J.; Neuhoff, M.T.; Hoyler, T.; Noir, E.; Tessier, C.; Sarret, S.; Thorsen, T.N.; Littlewood-Evans, A.; Zhang, J.; Hasan, M.; et al. TNF Leads to MtDNA Release and CGAS/STING-Dependent Interferon Responses That Support Inflammatory Arthritis. Cell Rep. 2021, 37, 109977. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current Pathogenesis and Therapeutic Options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Zernecke, A.; Weber, C. Chemokines in Atherosclerosis: Proceedings Resumed. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 742–750. [Google Scholar] [CrossRef]
- Koenen, R.R.; Weber, C. Therapeutic Targeting of Chemokine Interactions in Atherosclerosis. Nat. Rev. Drug Discov. 2010, 9, 141–153. [Google Scholar] [CrossRef]
- Opdenakker, G.; van Damme, J. Cytokines and Proteases in Invasive Processes: Molecular Similarities between Inflammation and Cancer. Cytokine 1992, 4, 251–258. [Google Scholar] [CrossRef]
- Korbecki, J.; Grochans, S.; Gutowska, I.; Barczak, K.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of Receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 Ligands. Int. J. Mol. Sci. 2020, 21, 7619. [Google Scholar] [CrossRef]
- Yoshimura, T. The Chemokine MCP-1 (CCL2) in the Host Interaction with Cancer: A Foe or Ally? Cell. Mol. Immunol. 2018, 15, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Strieter, R.M.; Burdick, M.D.; Gomperts, B.N.; Belperio, J.A.; Keane, M.P. CXC Chemokines in Angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 593–609. [Google Scholar] [CrossRef] [Green Version]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. CXC Chemokines in Cancer Angiogenesis and Metastases. Adv. Cancer Res. 2010, 106, 91–111. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Sun, S.; Li, Y.; Li, X.; Li, Z.; Liang, H. Identification of Therapeutic Targets and Prognostic Biomarkers Among CXC Chemokines in the Renal Cell Carcinoma Microenvironment. Front. Oncol. 2020, 9, 1555. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Frangogiannis, N.G. Chemokines in Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 35–52. [Google Scholar] [CrossRef]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A New Class of Membrane-Bound Chemokine with a CX3C Motif. Nature 1997, 385, 640–642. [Google Scholar] [CrossRef]
- Skoda, M.; Stangret, A.; Szukiewicz, D. Fractalkine and Placental Growth Factor: A Duet of Inflammation and Angiogenesis in Cardiovascular Disorders. Cytokine Growth Factor Rev. 2018, 39, 116–123. [Google Scholar] [CrossRef]
- Tian, Y.; Terkawi, M.A.; Onodera, T.; Alhasan, H.; Matsumae, G.; Takahashi, D.; Hamasaki, M.; Ebata, T.; Aly, M.K.; Kida, H.; et al. Blockade of XCL1/Lymphotactin Ameliorates Severity of Periprosthetic Osteolysis Triggered by Polyethylene-Particles. Front. Immunol. 2020, 11, 1720. [Google Scholar] [CrossRef]
- York, A.G.; Williams, K.J.; Argus, J.P.; Zhou, Q.D.; Brar, G.; Vergnes, L.; Gray, E.E.; Zhen, A.; Wu, N.C.; Yamada, D.H.; et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 2015, 163, 1716–1729. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.D.; Chi, X.; Lee, M.S.; Hsieh, W.Y.; Mkrtchyan, J.J.; Feng, A.C.; He, C.; York, A.G.; Bui, V.L.; Kronenberger, E.B.; et al. Interferon-Mediated Reprogramming of Membrane Cholesterol to Evade Bacterial Toxins. Nat. Immunol. 2020, 21, 746–755. [Google Scholar] [CrossRef]
- Gold, E.S.; Diercks, A.H.; Podolsky, I.; Podyminogin, R.L.; Askovich, P.S.; Treuting, P.M.; Aderem, A. 25-Hydroxycholesterol Acts as an Amplifier of Inflammatory Signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 10666–10671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, E.S.; Ramsey, S.A.; Sartain, M.J.; Selinummi, J.; Podolsky, I.; Rodriguez, D.J.; Moritz, R.L.; Aderem, A. ATF3 Protects against Atherosclerosis by Suppressing 25-Hydroxycholesterol-Induced Lipid Body Formation. J. Exp. Med. 2012, 209, 807–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Gage, J.; Hasu, M.; Thabet, M.; Whitman, S.C. Caspase-1 Deficiency Decreases Atherosclerosis in Apolipoprotein E-Null Mice. Can. J. Cardiol. 2012, 28, 222–229. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; La Bastide-Van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [Green Version]
- Cornicelli, J.A.; Butteiger, D.; Rateri, D.L.; Welch, K.; Daugherty, A. Interleukin-4 Augments Acetylated LDL-Induced Cholesterol Esterification in Macrophages. J. Lipid Res. 2000, 41, 376–383. [Google Scholar] [CrossRef]
- George, J.; Mulkins, M.; Shaish, A.; Casey, S.; Schatzman, R.; Sigal, E.; Harats, D. Interleukin (IL)-4 Deficiency Does Not Influence Fatty Streak Formation in C57BL/6 Mice. Atherosclerosis 2000, 153, 403–411. [Google Scholar] [CrossRef]
- Sämpi, M.; Ukkola, O.; Päivänsalo, M.; Kesäniemi, Y.A.; Binder, C.J.; Hörkkö, S. Plasma Interleukin-5 Levels Are Related to Antibodies Binding to Oxidized Low-Density Lipoprotein and to Decreased Subclinical Atherosclerosis. J. Am. Coll. Cardiol. 2008, 52, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Silveira, A.; McLeod, O.; Strawbridge, R.J.; Gertow, K.; Sennblad, B.; Baldassarre, D.; Veglia, F.; Deleskog, A.; Persson, J.; Leander, K.; et al. Plasma IL-5 Concentration and Subclinical Carotid Atherosclerosis. Atherosclerosis 2015, 239, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Ishigami, T.; Abe, K.; Aoki, I.; Minegishi, S.; Ryo, A.; Matsunaga, S.; Matsuoka, K.; Takeda, H.; Sawasaki, T.; Umemura, S.; et al. Anti-Interleukin-5 and Multiple Autoantibodies Are Associated with Human Atherosclerotic Diseases and Serum Interleukin-5 Levels. FASEB J. 2013, 27, 3437–3445. [Google Scholar] [CrossRef]
- Zhao, W.; Lei, T.; Li, H.; Sun, D.; Mo, X.; Wang, Z.; Zhang, K.; Ou, H. Macrophage-Specific Overexpression of Interleukin-5 Attenuates Atherosclerosis in LDL Receptor-Deficient Mice. Gene Ther. 2015, 22, 645–652. [Google Scholar] [CrossRef]
- Ren, W.; Wang, Z.; Wang, J.; Wu, Z.; Ren, Q.; Yu, A.; Ruan, Y. IL-5 Overexpression Attenuates Aortic Dissection by Reducing Inflammation and Smooth Muscle Cell Apoptosis. Life Sci. 2020, 241, 117144. [Google Scholar] [CrossRef]
- Chen, Y.; Duan, Y.; Kangs, Y.; Yang, X.; Jiang, M.; Zhangs, L.; Li, G.; Yin, Z.; Hu, W.; Dong, P.; et al. Activation of Liver X Receptor Induces Macrophage Interleukin-5 Expression. J. Biol. Chem. 2012, 287, 43340–43350. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhao, Z.; Wang, G.; Zou, J.; Yu, X.; Zhang, D.; Zeng, G.; Tang, C. Interleukin-5 Promotes ATP-Binding Cassette Transporter A1 Expression through MiR-211/JAK2/STAT3 Pathways in THP-1-Dervied Macrophages. Acta Biochim. Biophys. Sin. 2020, 52, 832–841. [Google Scholar] [CrossRef]
- Hartman, J.; Frishman, W.H. Inflammation and Atherosclerosis: A Review of the Role of Interleukin-6 in the Development of Atherosclerosis and the Potential for Targeted Drug Therapy. Cardiol. Rev. 2014, 22, 147–151. [Google Scholar] [CrossRef]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream to Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Cainzos-Achirica, M.; Enjuanes, C.; Greenland, P.; McEvoy, J.W.; Cushman, M.; Dardari, Z.; Nasir, K.; Budoff, M.J.; Al-Mallah, M.H.; Yeboah, J.; et al. The Prognostic Value of Interleukin 6 in Multiple Chronic Diseases and All-Cause Death: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2018, 278, 217–225. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Goldstein, D.R. Ageing and Atherosclerosis: Vascular Intrinsic and Extrinsic Factors and Potential Role of IL-6. Nat Rev. Cardiol. 2021, 18, 58–68. [Google Scholar] [CrossRef]
- López-Mejías, R.; González-Gay, M.A. IL-6: Linking Chronic Inflammation and Vascular Calcification. Nat. Rev. Rheumatol. 2019, 15, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; Abraham, N. Interleukin-7 Receptor Alpha in Innate Lymphoid Cells: More Than a Marker. Front. Immunol. 2019, 10, 2897. [Google Scholar] [CrossRef] [PubMed]
- Markina, Y.V.; Kirichenko, T.V.; Markin, A.M.; Yudina, I.Y.; Starodubova, A.V.; Sobenin, I.A.; Orekhov, A.N. Atheroprotective Effects of Glycyrrhiza glabra L. Molecules 2022, 27, 4697. [Google Scholar] [CrossRef]
- Arya, A.K.; Tripathi, R.; Kumar, S.; Tripathi, K. Recent Advances on the Association of Apoptosis in Chronic Non Healing Diabetic Wound. World J. Diabetes 2014, 5, 756. [Google Scholar] [CrossRef] [PubMed]
- Tamarit, B.; Bugault, F.; Pillet, A.H.; Lavergne, V.; Bochet, P.; Garin, N.; Schwarz, U.; Thèze, J.; Rose, T. Correction: Membrane Microdomains and Cytoskeleton Organization Shape and Regulate the IL-7 Receptor Signalosome in Human CD4 T-Cells. J. Biol. Chem. 2019, 294, 5212. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Paul, A.; Ko, K.W.S.; Sheldon, M.; Rich, B.E.; Terashima, T.; Dieker, C.; Cormier, S.; Li, L.; Nour, E.A.; et al. Interleukin-7 Induces Recruitment of Monocytes/Macrophages to Endothelium. Eur. Heart J. 2012, 33, 3114–3123. [Google Scholar] [CrossRef]
- Coulson, D.J.; Bakhashab, S.; Latief, J.S.; Weaver, J.U. MiR-126, IL-7, CXCR1/2 Receptors, Inflammation and Circulating Endothelial Progenitor Cells: The Study on Targets for Treatment Pathways in a Model of Subclinical Cardiovascular Disease (Type 1 Diabetes Mellitus). J. Transl. Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- An, Z.; Li, J.; Yu, J.; Wang, X.; Gao, H.; Zhang, W.; Wei, Z.; Zhang, J.; Zhang, Y.; Zhao, J.; et al. Neutrophil Extracellular Traps Induced by IL-8 Aggravate Atherosclerosis via Activation NF-ΚB Signaling in Macrophages. Cell Cycle 2019, 18, 2928–2938. [Google Scholar] [CrossRef]
- Tang, X.E.; Li, H.; Chen, L.Y.; Xia, X.D.; Zhao, Z.W.; Zheng, X.L.; Zhao, G.J.; Tang, C.K. IL-8 Negatively Regulates ABCA1 Expression and Cholesterol Efflux via Upregulating MiR-183 in THP-1 Macrophage-Derived Foam Cells. Cytokine 2019, 122, 154385. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Z.; Zhou, L. Interleukin 8 Inhibition Enhanced Cholesterol Efflux in Acetylated Low-Density Lipoprotein-Stimulated THP-1 Macrophages. J. Investig. Med. 2014, 62, 615–620. [Google Scholar] [CrossRef]
- Han, X.; Boisvert, W.A. Interleukin-10 Protects against Atherosclerosis by Modulating Multiple Atherogenic Macrophage Function. Thromb. Haemost. 2015, 113, 505–512. [Google Scholar] [CrossRef]
- Han, X.; Kitamoto, S.; Wang, H.; Boisvert, W.A. Interleukin-10 Overexpression in Macrophages Suppresses Atherosclerosis in Hyperlipidemic Mice. FASEB J. 2010, 24, 2869–2880. [Google Scholar] [CrossRef] [Green Version]
- Halvorsen, B.; Holm, S.; Yndestad, A.; Scholz, H.; Sagen, E.L.; Nebb, H.; Holven, K.B.; Dahl, T.B.; Aukrust, P. Interleukin-10 Increases Reverse Cholesterol Transport in Macrophages through Its Bidirectional Interaction with Liver X Receptor α. Biochem. Biophys. Res. Commun. 2014, 450, 1525–1530. [Google Scholar] [CrossRef]
- Stöger, J.L.; Boshuizen, M.C.S.; Brufau, G.; Gijbels, M.J.J.; Wolfs, I.M.J.; van der Velden, S.; Pöttgens, C.C.H.; Vergouwe, M.N.; Wijnands, E.; Beckers, L.; et al. Deleting Myeloid IL-10 Receptor Signalling Attenuates Atherosclerosis in LDLR−/− Mice by Altering Intestinal Cholesterol Fluxes. Thromb. Haemost. 2016, 116, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Hauer, A.D.; Uyttenhove, C.; de Vos, P.; Stroobant, V.; Renauld, J.C.; van Berkel, U.C.; van Snick, J.; Kuiper, J. Blockade of Interleukin-12 Function by Protein Vaccination Attenuates Atherosclerosis. Circulation 2005, 112, 1054–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, K.Y.; Clanton, E.C.; Gehrig, J.L.; Granger, D.N. Role of Interleukin 12 in Hypercholesterolemia-Induced Inflammation. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2623–H2629. [Google Scholar] [CrossRef] [Green Version]
- Low, L.D.; Lu, L.; Chan, C.Y.; Chen, J.; Yang, H.H.; Yu, H.; Lee, C.G.L.; Ng, K.H.; Yap, H.K. IL-13-Driven Alterations in Hepatic Cholesterol Handling Contributes to Hypercholesterolemia in a Rat Model of Minimal Change Disease. Clin. Sci. 2020, 134, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liu, M.-F.; Huang, J.-N.; Li, J.-M.; Jiang, J.; Wang, J.-A. Role of Interleukin-15 in Cardiovascular Diseases. J. Cell Mol. Med. 2020, 24, 7094–7101. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, B.; Schwimmbeck, P.L.; Haas, C.S.; Lindemann, S. Effect of Interleukin-15 on the Course of Myocarditis in Coxsackievirus B3-Infected BALB/c Mice. Can. J. Cardiol. 2009, 25, e248. [Google Scholar] [CrossRef] [Green Version]
- Yeghiazarians, Y.; Honbo, N.; Imhof, I.; Woods, B.; Aguilera, V.; Ye, J.; Boyle, A.J.; Karliner, J.S. IL-15: A Novel pro-Survival Signaling Pathway in Cardiomyocytes. J. Cardiovasc. Pharmacol. 2014, 63, 406. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Liu, Z.; Liu, J.; Zhou, P.; Liu, Y.; Lu, X. The Paradoxical Role of IL-17 in Atherosclerosis. Cell Immunol. 2015, 297, 33–39. [Google Scholar] [CrossRef]
- Allam, G.; Abdel-Moneim, A.; Gaber, A.M. The Pleiotropic Role of Interleukin-17 in Atherosclerosis. Biomed. Pharmacother. 2018, 106, 1412–1418. [Google Scholar] [CrossRef]
- Kidani, Y.; Bensinger, S.J. Reviewing the Impact of Lipid Synthetic Flux on Th17 Function. Curr. Opin. Immunol. 2017, 46, 121–126. [Google Scholar] [CrossRef]
- Ray, M.; Autieri, M.V. Regulation of Pro- and Anti-Atherogenic Cytokines. Cytokine 2019, 122, 154175. [Google Scholar] [CrossRef]
- Xia, Q.; Xiang, X.; Patel, S.; Puranik, R.; Xie, Q.; Bao, S. Characterisation of IL-22 and Interferon-Gamma-Inducible Chemokines in Human Carotid Plaque. Int. J. Cardiol. 2012, 154, 187–189. [Google Scholar] [CrossRef]
- Rattik, S.; Hultman, K.; Rauch, U.; Söderberg, I.; Sundius, L.; Ljungcrantz, I.; Hultgårdh-Nilsson, A.; Wigren, M.; Björkbacka, H.; Fredrikson, G.N.; et al. IL-22 Affects Smooth Muscle Cell Phenotype and Plaque Formation in Apolipoprotein E Knockout Mice. Atherosclerosis 2015, 242, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.W.; Hu, Y.; Liu, J.; Yang, H.; Huang, P. Interleukin-22: A Potential Therapeutic Target in Atherosclerosis. Mol. Med. 2021, 27, 88. [Google Scholar] [CrossRef]
- Saggini, A.; Anogeianaki, A.; Maccauro, G.; Teté, S.; Salini, V.; Caraffa, A.; Conti, F.; Fulcheri, M.; Galzio, R.; Shaik-Dasthagirisaheb, Y.B. Cholesterol, Cytokines and Diseases. Int. J. Immunopathol. Pharmacol. 2011, 24, 567–581. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Valdes-Marquez, E.; Hill, M.; Gordon, J.; Farrall, M.; Hamsten, A.; Watkins, H.; Hopewell, J.C. Plasma Cytokines and Risk of Coronary Heart Disease in the PROCARDIS Study. Open Heart 2018, 5, e000807. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, R.; Mu, H.; Wang, X.; Yao, Q.; Chen, C. Reverse Cholesterol Transport and Cholesterol Efflux in Atherosclerosis. QJM 2005, 98, 845–856. [Google Scholar] [CrossRef]
- Hashizume, M.; Mihara, M. Atherogenic Effects of TNF-α and IL-6 via up-Regulation of Scavenger Receptors. Cytokine 2012, 58, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Kirkiles-Smith, N.C.; McNiff, J.M.; Pober, J.S. TRAIL Induces Apoptosis and Inflammatory Gene Expression in Human Endothelial Cells. J. Immunol. 2003, 171, 1526–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bartolo, B.A.; Chan, J.; Bennett, M.R.; Cartland, S.; Bao, S.; Tuch, B.E.; Kavurma, M.M. TNF-Related Apoptosis-Inducing Ligand (TRAIL) Protects against Diabetes and Atherosclerosis in Apoe −/− Mice. Diabetologia 2011, 54, 3157–3167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Bartolo, B.A.; Cartland, S.P.; Genner, S.; Manuneedhi Cholan, P.; Vellozzi, M.; Rye, K.A.; Kavurma, M.M. HDL Improves Cholesterol and Glucose Homeostasis and Reduces Atherosclerosis in Diabetes-Associated Atherosclerosis. J. Diabetes Res. 2021, 2021, 6668506. [Google Scholar] [CrossRef] [PubMed]
- Cartland, S.P.; Harith, H.H.; Genner, S.W.; Dang, L.; Cogger, V.C.; Vellozzi, M.; Di Bartolo, B.A.; Thomas, S.R.; Adams, L.A.; Kavurma, M.M. Non-Alcoholic Fatty Liver Disease, Vascular Inflammation and Insulin Resistance Are Exacerbated by TRAIL Deletion in Mice. Sci. Rep. 2017, 7, 1898. [Google Scholar] [CrossRef] [Green Version]
- Chyuan, I.T.; Tsai, H.F.; Wu, C.S.; Hsu, P.N. TRAIL Suppresses Gut Inflammation and Inhibits Colitogeic T-Cell Activation in Experimental Colitis via an Apoptosis-Independent Pathway. Mucosal. Immunol. 2019, 12, 980–989. [Google Scholar] [CrossRef] [Green Version]
- Zoller, V.; Funcke, J.B.; Roos, J.; Dahlhaus, M.; Abd El Hay, M.; Holzmann, K.; Marienfeld, R.; Kietzmann, T.; Debatin, K.M.; Wabitsch, M.; et al. Trail (TNF-Related Apoptosis-Inducing Ligand) Induces an Inflammatory Response in Human Adipocytes. Sci. Rep. 2017, 7, 5691. [Google Scholar] [CrossRef] [Green Version]
- Manuneedhi Cholan, P.; Cartland, S.P.; Dang, L.; Rayner, B.S.; Patel, S.; Thomas, S.R.; Kavurma, M.M. TRAIL Protects against Endothelial Dysfunction in Vivo and Inhibits Angiotensin-II-Induced Oxidative Stress in Vascular Endothelial Cells in Vitro. Free Radic. Biol. Med. 2018, 126, 341–349. [Google Scholar] [CrossRef]
- Volpato, S.; Ferrucci, L.; Secchiero, P.; Corallini, F.; Zuliani, G.; Fellin, R.; Guralnik, J.M.; Bandinelli, S.; Zauli, G. Association of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand with Total and Cardiovascular Mortality in Older Adults. Atherosclerosis 2011, 215, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of CC-Chemokines in the Regulation of Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1856. [Google Scholar] [CrossRef] [Green Version]
- Georgakis, M.K.; Bernhagen, J.; Heitman, L.H.; Weber, C.; Dichgans, M. Targeting the CCL2-CCR2 Axis for Atheroprotection. Eur. Heart J. 2022, 43, 1799–1808. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, N.S.; Ying, R.; Xie, Y.; Chen, J.Y.; Wang, X.Q.; Gu, Z.J.; Mai, J.T.; Liu, W.H.; Wu, M.X.; et al. Macrophage-Derived Foam Cells Impair Endothelial Barrier Function by Inducing Endothelial-Mesenchymal Transition via CCL-4. Int. J. Mol. Med. 2017, 40, 558. [Google Scholar] [CrossRef] [Green Version]
- Vila-Caballer, M.; González-Granado, J.M.; Zorita, V.; Abu Nabah, Y.N.; Silvestre-Roig, C.; del Monte-Monge, A.; Molina-Sánchez, P.; Ait-Oufella, H.; Andrés-Manzano, M.J.; Sanz, M.J.; et al. Disruption of the CCL1-CCR8 Axis Inhibits Vascular Treg Recruitment and Function and Promotes Atherosclerosis in Mice. J. Mol. Cell. Cardiol. 2019, 132, 154–163. [Google Scholar] [CrossRef]
- Benna, M.; Guy, J.B.; Bosacki, C.; Jmour, O.; Ben Mrad, M.; Ogorodniitchouk, O.; Soltani, S.; Lan, M.; Daguenet, E.; Mery, B.; et al. Chemoradiation and Granulocyte-Colony or Granulocyte Macrophage-Colony Stimulating Factors (G-CSF or GM-CSF): Time to Think out of the Box? Br. J. Radiol. 2020, 93, 20190147. [Google Scholar] [CrossRef]
- Lappalainen, J.; Yeung, N.; Nguyen, S.D.; Jauhiainen, M.; Kovanen, P.T.; Lee-Rueckert, M. Cholesterol Loading Suppresses the Atheroinflammatory Gene Polarization of Human Macrophages Induced by Colony Stimulating Factors. Sci. Rep. 2021, 11, 4923. [Google Scholar] [CrossRef]
- Prud’homme, G.J. Pathobiology of Transforming Growth Factor Beta in Cancer, Fibrosis and Immunologic Disease, and Therapeutic Considerations. Lab. Investig. 2007, 87, 1077–1091. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.Y.; Qin, L.; Tellides, G.; Simons, M. Fibroblast Growth Factor Receptor 1 Is a Key Inhibitor of TGFβ Signaling in the Endothelium. Sci. Signal. 2014, 7, ra90. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β Signalling Drives Vascular Inflammation and Atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, J.; Jackson, A.O.; Zhu, X.; Chen, H.; Chen, W.; Gui, Q.; Yin, K. Apolipoprotein A1 Inhibits the TGF-Β1-Induced Endothelial-to-Mesenchymal Transition of Human Coronary Artery Endothelial Cells. Cardiology 2017, 137, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, F.; Ahmad, Z.; Thomas, R.; Melhem, M.; Snider, A.J.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; Ahmad, R. Neutral Sphingomyelinase 2 Regulates Inflammatory Responses in Monocytes/Macrophages Induced by TNF-α. Sci. Rep. 2020, 10, 16802. [Google Scholar] [CrossRef]
- Saikh, K.U.; Khan, A.S.; Kissner, T.; Ulrich, R.G. IL-15-Induced Conversion of Monocytes to Mature Dendritic Cells. Clin. Exp. Immunol. 2001, 126, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Mitsis, A.; Kadoglou, N.P.E.; Lambadiari, V.; Alexiou, S.; Theodoropoulos, K.C.; Avraamides, P.; Kassimis, G. Prognostic Role of Inflammatory Cytokines and Novel Adipokines in Acute Myocardial Infarction: An Updated and Comprehensive Review. Cytokine 2022, 153, 155848. [Google Scholar] [CrossRef]
- Jiang, Y.; Yu, M.; Hu, X.; Han, L.; Yang, K.; Ba, H.; Zhang, Z.; Yin, B.; Yang, X.P.; Li, Z.; et al. STAT1 Mediates Transmembrane TNF-Alpha-Induced Formation of Death-Inducing Signaling Complex and Apoptotic Signaling via TNFR1. Cell Death Differ. 2017, 24, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Evensen, Ø.; Munang’andu, H.M. Transcriptome Analysis Shows That IFN-I Treatment and Concurrent SAV3 Infection Enriches MHC-I Antigen Processing and Presentation Pathways in Atlantic Salmon-Derived Macrophage/Dendritic Cells. Viruses 2019, 11, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabin, R.L.; Walter, M.R. Editorial: Structures, Signaling Mechanisms, and Functions of Types I and III Interferons. Front. Immunol. 2021, 12, 638479. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, Regulation, and Involvement in Disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef] [PubMed]
- Munjal, A.; Khandia, R. Atherosclerosis: Orchestrating Cells and Biomolecules Involved in Its Activation and Inhibition. Adv. Protein Chem. Struct. Biol. 2020, 120, 85–122. [Google Scholar] [CrossRef]
- Janssens, R.; Struyf, S.; Proost, P. The Unique Structural and Functional Features of CXCL12. Cell Mol. Immunol. 2018, 15, 299. [Google Scholar] [CrossRef] [Green Version]
- Mossadegh-Keller, N.; Gentek, R.; Gimenez, G.; Bigot, S.; Mailfert, S.; Sieweke, M.H. Developmental Origin and Maintenance of Distinct Testicular Macrophage Populations. J. Exp. Med. 2017, 214, 2829–2841. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, C.C. Inflammatory Response of Macrophages in Infection. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 138–152. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β Induces M2-like Macrophage Polarization via SNAILmediated Suppression of a pro-Inflammatory Phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Bradwin, G.; Hasan, A.A.; Rifai, N. Comparison of Interleukin-6, C-Reactive Protein, and Low-Density Lipoprotein Cholesterol as Biomarkers of Residual Risk in Contemporary Practice: Secondary Analyses from the Cardiovascular Inflammation Reduction Trial. Eur. Heart J. 2020, 41, 2952–2961. [Google Scholar] [CrossRef] [Green Version]
- King, V.L.; Cassis, L.A.; Daugherty, A. Interleukin-4 Does Not Influence Development of Hypercholesterolemia or Angiotensin II-Induced Atherosclerotic Lesions in Mice. Am. J. Pathol. 2007, 171, 2040–2047. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Yang, S.; Xing, Y.; Pan, L.; Zhao, R.; Zhao, Y.; Liu, L.; Wu, M. Novel Insights and Current Evidence for Mechanisms of Atherosclerosis: Mitochondrial Dynamics as a Potential Therapeutic Target. Front. Cell Dev. Biol. 2021, 9, 673839. [Google Scholar] [CrossRef]
- Zhou, M.S.; Chadipiralla, K.; Mendez, A.J.; Jaimes, E.A.; Silverstein, R.L.; Webster, K.; Raij, L. Nicotine Potentiates Proatherogenic Effects of OxLDL by Stimulating and Upregulating Macrophage CD36 Signaling. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H563–H574. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Tsang, T.M.; Qiu, Y.; Dayrit, J.K.; Freij, J.B.; Huffnagle, G.B.; Olszewski, M.A. Macrophage M1/M2 Polarization Dynamically Adapts to Changes in Cytokine Microenvironments in Cryptococcus Neoformans Infection. mBio 2013, 4, e00264-13. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Kwak, A.; Jhun, H.; Lee, S.; Jo, S.; Lee, J.; Kang, T.B.; Her, E.; Bae, S.; Lee, Y.; et al. Development of an Interleukin (IL)-33 Sandwich ELISA Kit Specific for Mature IL-33. J. Immunoass. Immunochem. 2016, 37, 585–596. [Google Scholar] [CrossRef]
- Marković, I.; Savvides, S.N. Modulation of Signaling Mediated by TSLP and IL-7 in Inflammation, Autoimmune Diseases, and Cancer. Front. Immunol. 2020, 11, 1557. [Google Scholar] [CrossRef]
- Chen, S.; Xie, X.; Wang, Y.; Gao, Y.; Xie, X.; Yang, J.; Ye, J. Association between Leukocyte Mitochondrial DNA Content and Risk of Coronary Heart Disease: A Case-Control Study. Atherosclerosis 2014, 237, 220–226. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ley, K. Atherosclerosis: A Chronic Inflammatory Disease with an Autoimmune Component. Circ. Res. 2018, 123, 1118. [Google Scholar] [CrossRef] [PubMed]
- Engelbertsen, D.; Lichtman, A.H. Innate Lymphoid Cells in Atherosclerosis. Eur. J. Pharmacol. 2017, 816, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 Protects from Atherosclerosis and Modulates Plaque Composition by Skewing the Macrophage Phenotype. EMBO Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-Induced Cytokine Production in Human Monocytes and Macrophages. Crit. Rev. Immunol. 2011, 31, 379–446. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, J.; Liu, J.; Ye, J.; Xu, Y.; Wang, Z.; Yu, J.; Ye, D.; Zhao, M.; Feng, Y.; et al. The Role of Interleukin-10 Family Members in Cardiovascular Diseases. Int. Immunopharmacol. 2021, 94, 107475. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Group | Action Molecules | Secreting Cells | Effector Cells | Effects |
|---|---|---|---|---|
| TNF Super Family | TNF-a | Macrophages, lymphoid cells, mast cells, endothelial cells, cardiac myocytes, adipose tissue, fibroblasts and neurons. | Macrophages, monocytes, B cells, T cells, NK cells, endothelial cells. | Stimulates phagocytosis, production of IL-1 oxidants and the inflammatory lipid prostaglandin E2 (PGE2) [151], IL-10 production, proliferation, Ig production, HLA-DR and CD25 expression, GM-CSF production [152], enhancement of cytotoxic activity, cell death, induction of pro-coagulant agents [153], adhesion molecules and pro-inflammatory cytokines [154]. |
| Interferons | IFN-I | Fibroblasts and monocytes. | Natural killer cells and macrophages. | Activate immune cells, increase host defenses by up-regulating antigen presentation by virtue of increasing the expression of major histocompatibility complex (MHC) antigens [155]. |
| IFN-II | Adaptive immune cells, more specifically CD4+ T helper 1 (Th1) cells, natural killer (NK) cells, and CD8+ cytotoxic T cells. | Macrophages, B cells, CD8+ cytotoxic T cells. | Promote inflammation, antiviral or antibacterial activity, and cell proliferation and differentiation [156]. | |
| IFN-III | Type 2 myeloid dendritic cells. | Epithelial cells, neutrophils, B cells and dendritic cells. | Modulate the immune response after a pathogen has been sensed in the organism, their functions are mostly anti-viral and anti-proliferative [157]. | |
| Chemokines | CC | Cells of innate and adaptive immunity. | T cells, eosinophils and basophils, monocytes, NK cells and dendritic cells. | Induces monocytes to leave the bloodstream and enter the surrounding tissue to become tissue macrophages, induce the migration of monocytes and other cell types such as NK cells and dendritic cells [158]. |
| CXC | Neutrophils, lymphocytes. | Induces the migration of neutrophils, activating their metabolic and degranulation [159], chemoattractant for lymphocytes [160]. | ||
| C | T cell, dendritic cells. | Involved in cross-presentation, antigen uptake, and induction of innate as well as adaptive cytotoxic immunity, to increate T cells in joints that are affected with rheumatoid arthritis [76]. | ||
| CX3C | T cell, monocytes, leukocytes. | Soluble, potently chemoattracts T cells and monocytes [77], cell-bound chemokine promotes strong adhesion of leukocytes to activated endothelial cells. | ||
| CSF | CSF1 | Different types of cells. | Hematopoietic stem cells, monocytes, macrophages. | Causes hematopoietic stem cells to differentiate into macrophages or other related cell types [161]. |
| CSF2 | Macrophages, T cells, mast cells, natural killer cells, endothelial cells and fibroblasts. | Affects more cell types, especially macrophages and eosinophils. | Stimulates stem cells to produce granulocytes (neutrophils, eosinophils, basophils) and monocytes. Activates the maturation of monocytes and dendritic cells [152]. | |
| CSF3 | Endothelium, macrophages, and a number of other immune cells. | Precursor cells in the bone marrow, neutrophil precursors and mature neutrophils, hematopoietic stem cell. | Stimulates the bone marrow to produce granulocytes and stem cells and release them into the bloodstream, stimulates the survival, proliferation, differentiation, and function of neutrophil precursors and mature neutrophils. | |
| TGF | TGFα | Macrophages, brain cells and keratinocytes. | Epithelial cells, neural cell. | Induces epithelial development, stimulates neural cell proliferation [162]. |
| TGFβ | All white blood cell lineages. | Macrophages, stem cell, T cell, B cell. | Plays crucial roles in tissue regeneration, cell differentiation [149], embryonic development and regulation of the immune system [163]. | |
| IL-1 family | IL-1β | Activated macrophages. | Different cell types. | Important mediator of the inflammatory response [164], and is involved in a variety of cellular activities [94], including cell proliferation, differentiation and apoptosis [53]. |
| IL-4 | Mast cells, Th2 cells, eosinophils and basophils. | B cell and T cell, macrophages. | Induces differentiation of naive helper T cells (Th0 cells) to Th2 cells [165], promotes alternative activation of macrophages into M2 cells and inhibits classical activation of macrophages into M1 cells [166]. | |
| IL-5 | Type-2 T helper cells and mast cells. | B cell, eosinophils. | Stimulates B cell growth and increases immunoglobulin secretion, mediator in eosinophil activation. | |
| IL-6 | Macrophages, osteoblasts, smooth muscle cells. | Neutrophils, B cells, T cells. | Stimulating acute phase protein synthesis [167], as well as the production of neutrophils in the bone marrow [168], it supports the growth of B cells and is antagonistic to regulatory T cells. | |
| IL-7 | Stromal cells in the bone marrow and thymus, keratinocytes, dendritic cells, hepatocytes, neurons and epithelial cells. | B cells, T cells and NK cells. | Stimulates the differentiation of multipotent (pluripotent) hematopoietic stem cells into lymphoid progenitor cells [169], stimulates proliferation of all cells in the lymphoid lineage [170]. | |
| IL-8 is a member of the CXC family of chemokines | IL-8 | Macrophages and other cell types. | Granulocytes. | Induces chemotaxis in target cells, primarily neutrophils, stimulates phagocytosis, promoter of angiogenesis [171]. |
| IL-10 | Monocytes and, to a lesser extent, lymphocytes. | Th1, Macrophages, B cell. | It downregulates the expression of Th1 cytokines, MHC class II antigens, and co-stimulatory molecules on macrophages. It also enhances B cell survival, proliferation, and antibody production. IL-10 can block NF-κB activity, and is involved in the regulation of the JAK-STAT signaling pathway [172]. | |
| IL-12 family | IL-12 | Dendritic cells, macrophages, neutrophils, and human B- lymphoblastoid cells (NC-37). | T cells, NK cells. | Stimulates the growth and function of T cells [173], stimulates the production of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) from T cells and NK cells, and reduces IL-4 mediated suppression of IFN-γ, block the formation of new blood vessels [117]. |
| IL-13 | Th2 cells, CD4 cells, natural killer T cell, mast cells, basophils, eosinophils and nuocytes. | Hematopoietic cells, B cell. | Regulator in IgE synthesis, goblet cell hyperplasia, mucus hypersecretion, airway hyperresponsiveness, fibrosis and chitinase up-regulation [174]. | |
| IL-15 | Mononuclear phagocytes. | NK cells, T cells. | Induces the proliferation of natural killer cells [175]. | |
| IL17 family | IL-17A | Activated T cells. | Th17. | Regulates the activities of NF-kappaB and mitogen-activated protein kinases, stimulate the expression of IL6 and cyclooxygenase-2 (PTGS2/COX-2), as well as enhances the production of nitric oxide (NO). |
| IL-22 | Tissue cells, αβ T cells classes Th1, Th22 and Th17 along with γδ T cells, NKT, ILC3, neutrophils and macrophages. | Non-hematopoietic cells—mainly stromal and epithelial cells. | Stimulation of cell survival, proliferation and synthesis of antimicrobials [176]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markin, A.M.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Chakal, D.A.; Breshenkov, D.G.; Charchyan, E.R. The Role of Cytokines in Cholesterol Accumulation in Cells and Atherosclerosis Progression. Int. J. Mol. Sci. 2023, 24, 6426. https://doi.org/10.3390/ijms24076426
Markin AM, Markina YV, Bogatyreva AI, Tolstik TV, Chakal DA, Breshenkov DG, Charchyan ER. The Role of Cytokines in Cholesterol Accumulation in Cells and Atherosclerosis Progression. International Journal of Molecular Sciences. 2023; 24(7):6426. https://doi.org/10.3390/ijms24076426
Chicago/Turabian StyleMarkin, Alexander M., Yuliya V. Markina, Anastasia I. Bogatyreva, Taisiya V. Tolstik, Deyyara A. Chakal, Denis G. Breshenkov, and Eduard R. Charchyan. 2023. "The Role of Cytokines in Cholesterol Accumulation in Cells and Atherosclerosis Progression" International Journal of Molecular Sciences 24, no. 7: 6426. https://doi.org/10.3390/ijms24076426