
The Landscape of Accessible Chromatin and Developmental Transcriptome Maps Reveal a Genetic Mechanism of Skeletal Muscle Development in Pigs
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
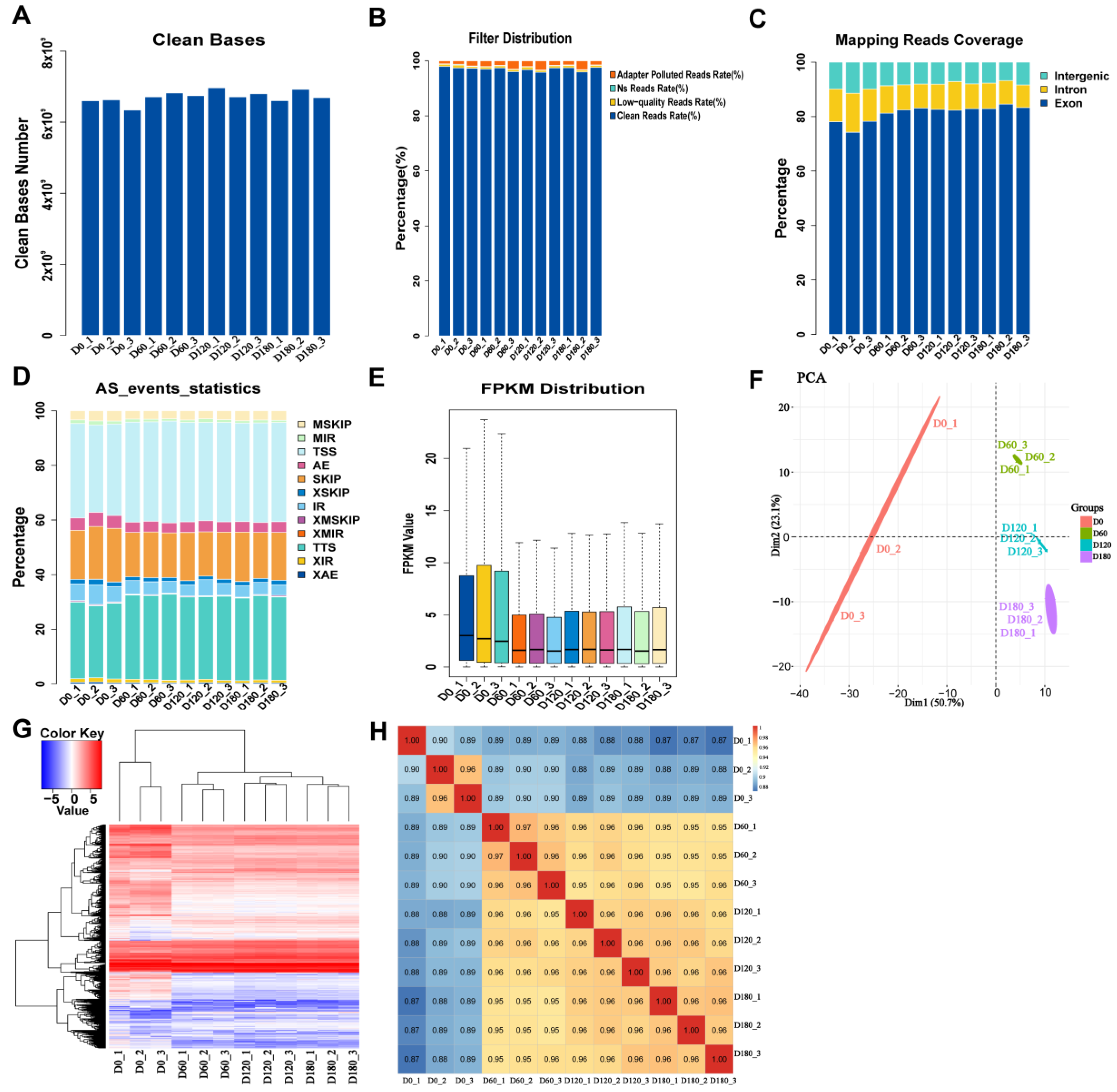
2.1. Data Filtering, Alignment Analysis, and Expression Statistics for RNA-Seq
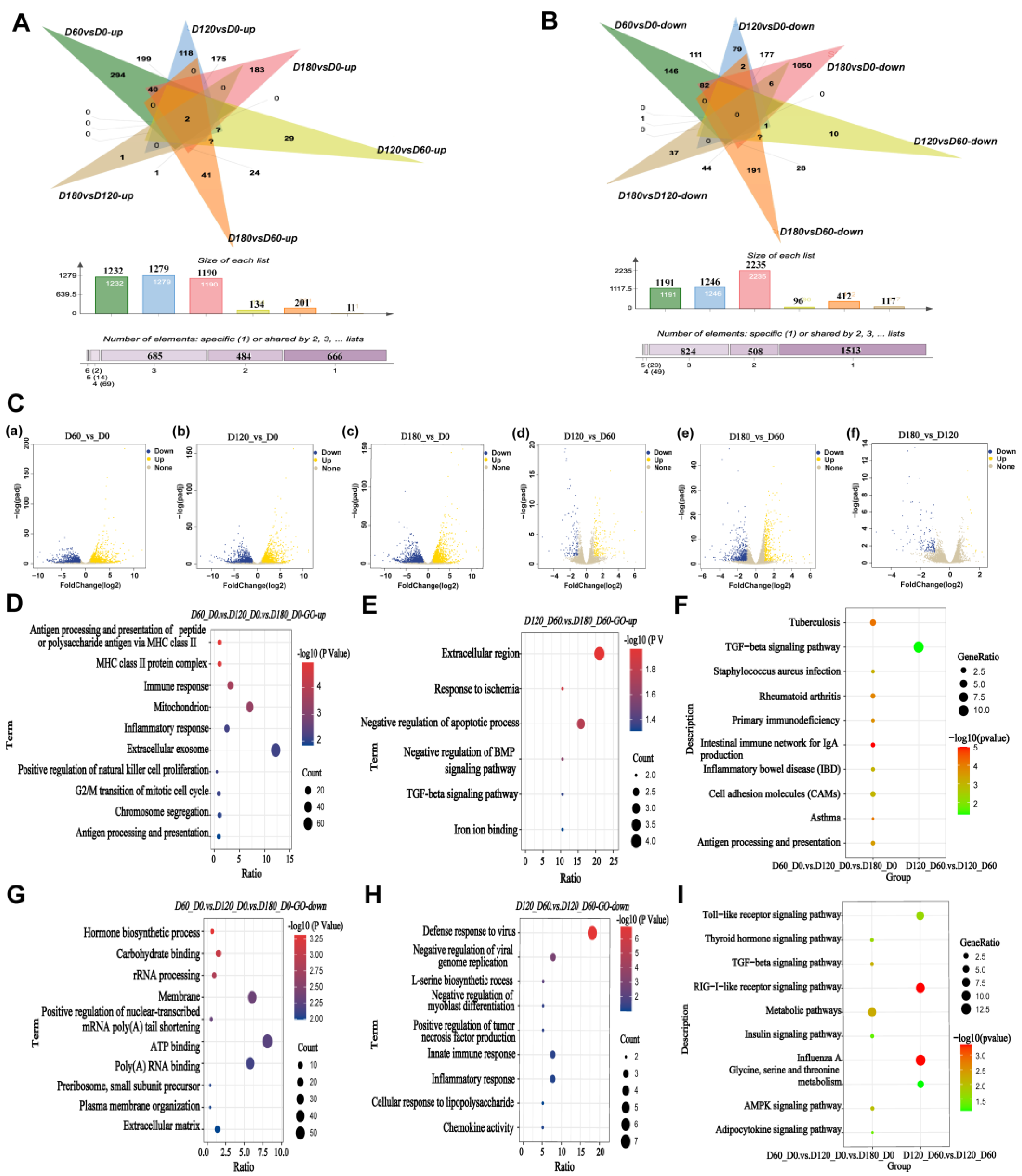
2.2. Enrichment Analysis of Differentially Expressed Genes during Postnatal Skeletal Muscle Growth
2.3. Trend Analysis for All RNA-Seq Genes, Cluster Analysis, and Gene Ontology
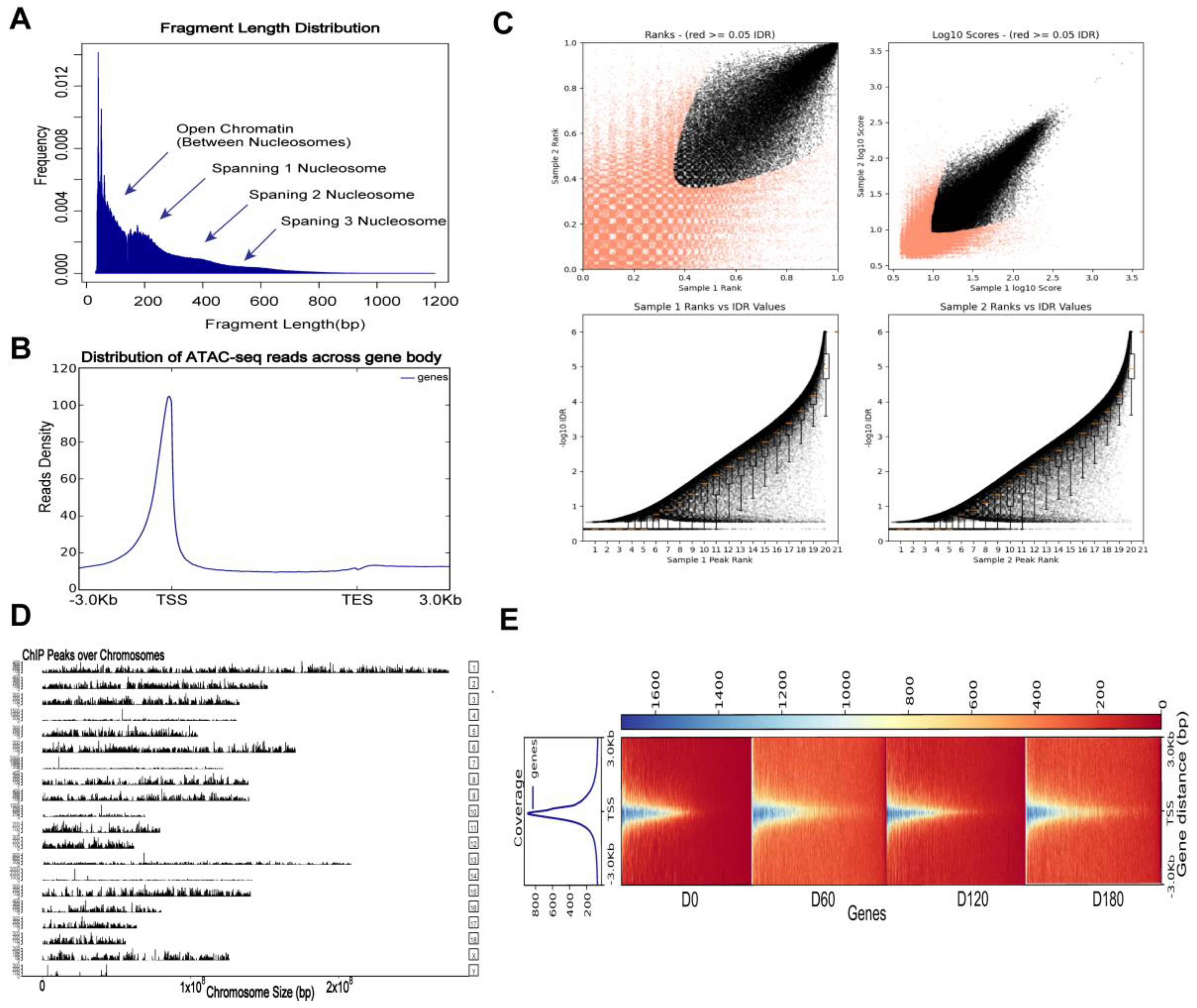
2.4. ATAC-Seq Read Mapping, Processing, and Visualization
2.5. Enrichment Analysis of nearby Genes of Differential Open Chromatin Regions
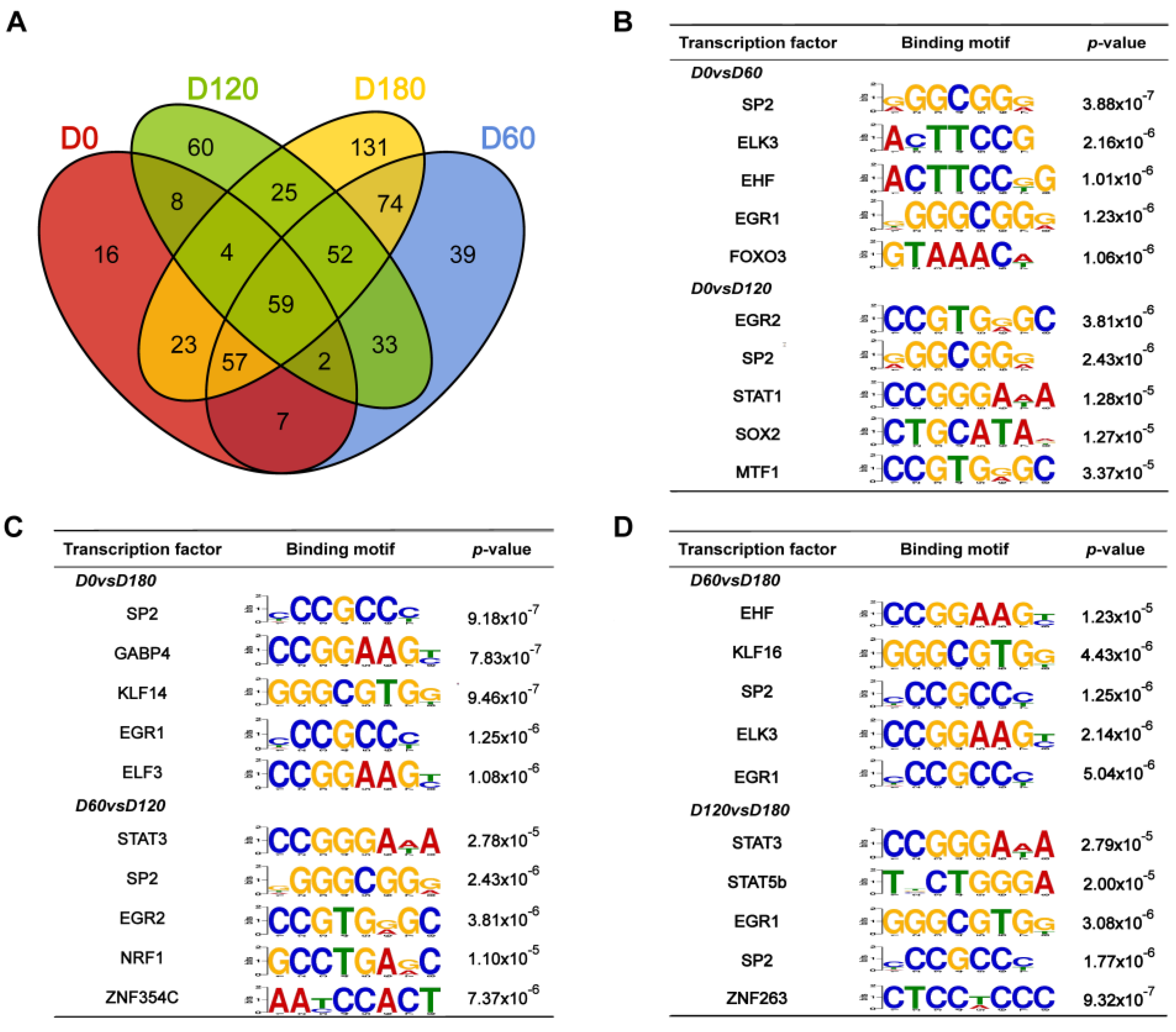
2.6. Motif Analysis of Differential Peaks during Postnatal Porcine Skeletal Muscle Development
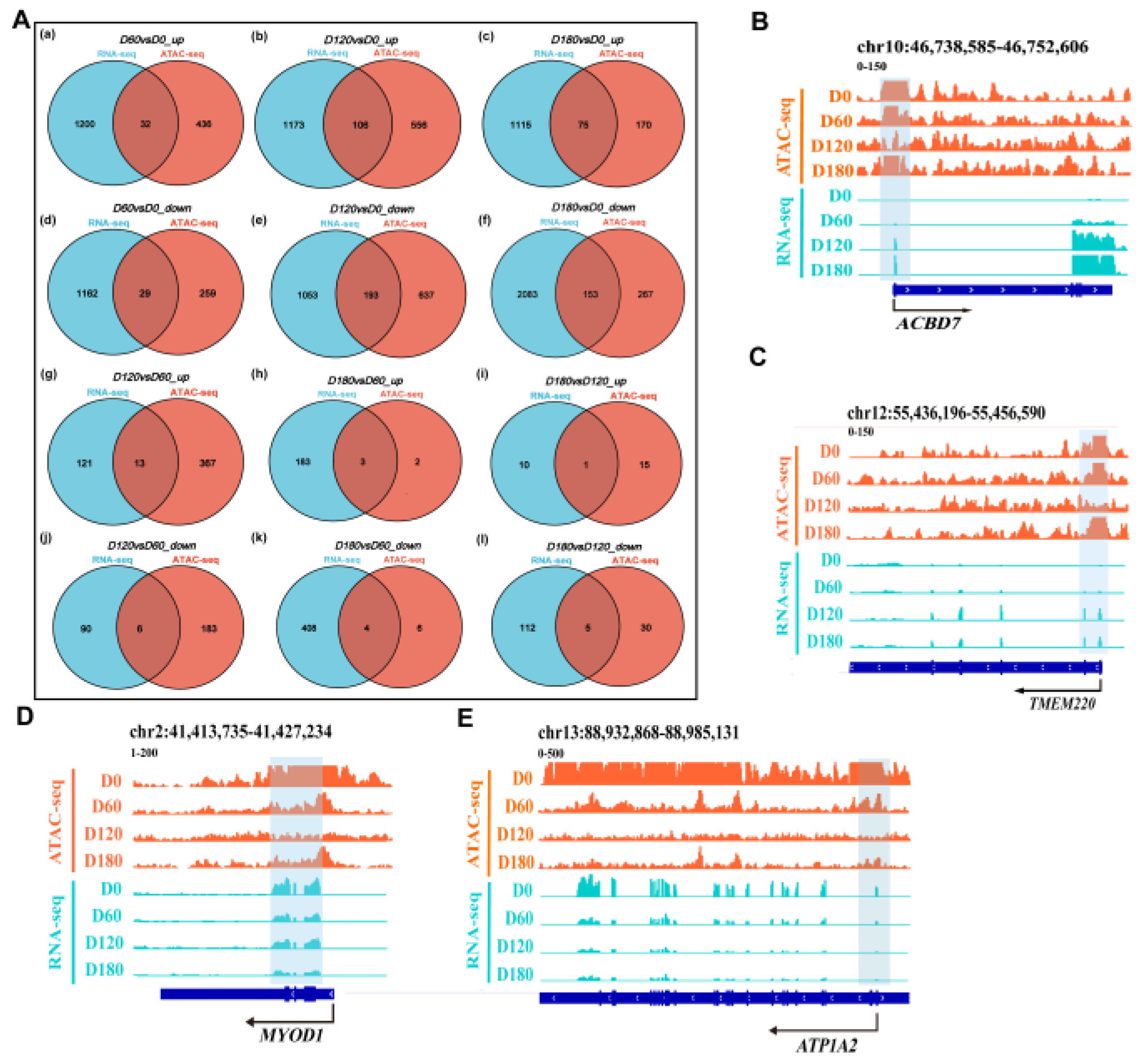
2.7. Relationship between Chromatin Accessibility and Gene Expression in Muscle Tissues
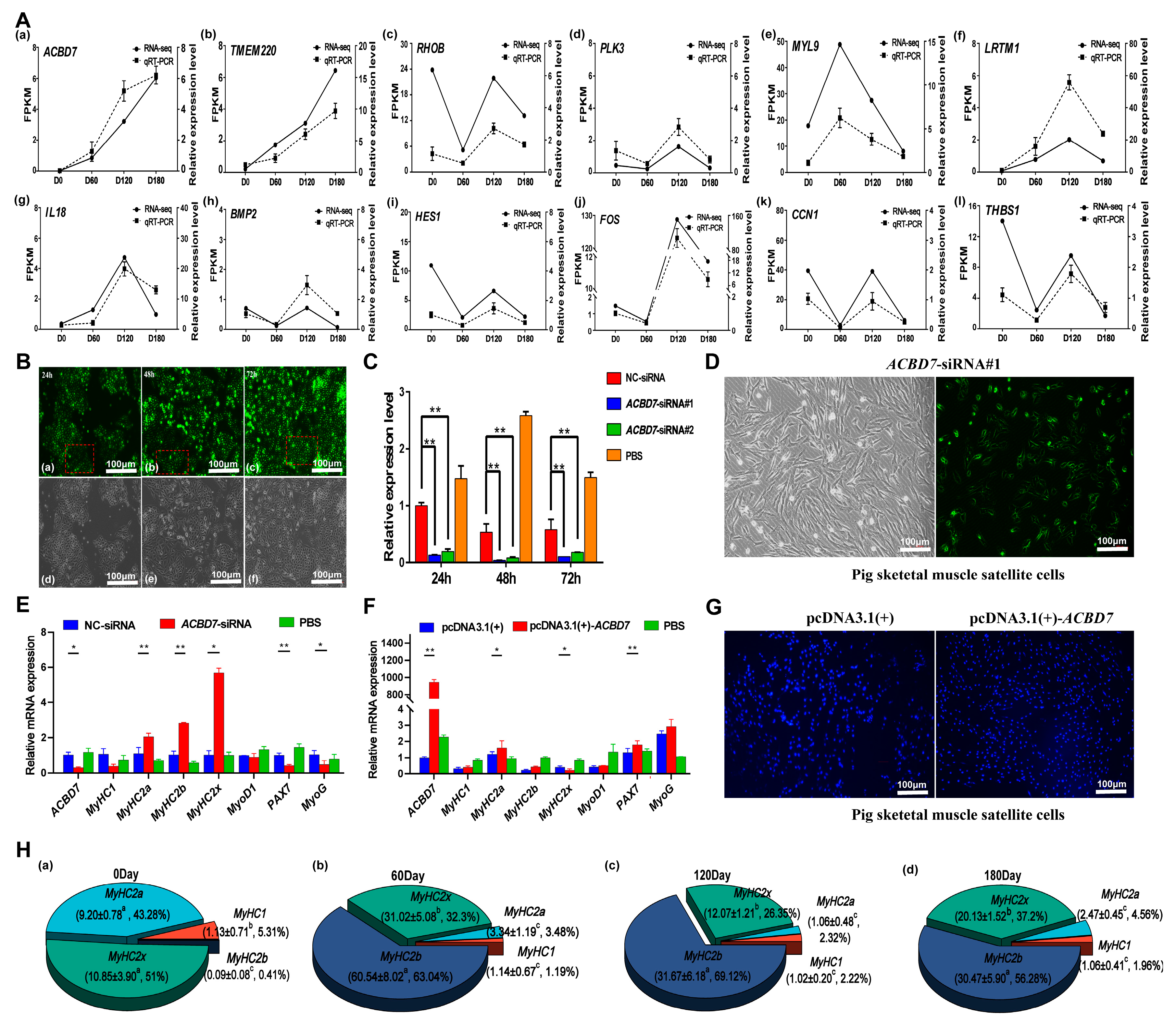
2.8. Validation of Screened Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animal Samples
4.3. RNA Extraction, Library Construction, and RNA Sequencing
4.4. Alignment of Reads to the Reference Genome for Transcript Assembly and Gene Expression Analysis
4.5. Assay and Library Preparation of Transposase Accessible Chromatin (ATAC)
4.6. ATAC-Seq Library Quality Inspection and Sequencing Data Quality Control and Peak Calling
4.7. Mapping and Normalization of ATAC-Seq
4.8. Analysis of Differential Chromatin Accessibility
4.9. Real-Time Fluorescent Quantitative Polymerase Chain Reaction (qRT-PCR)
4.10. Pathway Enrichment Analysis
4.11. Cell Culture
4.12. Transwell Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, K.; Baxter, T.; Muir, W.M.; Groenen, M.A.; Schook, L.B. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int. J. Biol. Sci. 2007, 3, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunney, J.K.; Van Goor, A.; Walker, K.E.; Hailstock, T.; Franklin, J.; Dai, C. Importance of the pig as a human biomedical model. Sci. Transl. Med. 2021, 13, eabd5758. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Wang, Y.; Liu, B.; Yin, F. Hypoglycemic drug liraglutide alleviates low muscle mass by inhibiting the expression of MuRF1 and MAFbx in diabetic muscle atrophy. J. Chin. Med. Assoc. 2023, 86, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhao, P.; Ren, Z. Construction of a Porcine Skeletal Muscle-Specific Promoter by Inducing the Seed Region of miR-208a. Mol. Biotechnol. 2022, 64, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Chal, J.; Pourquie, O. Making muscle, skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, K.; Corona, B.T.; Walters, T.J. Therapeutic strategies for preventing skeletal muscle fibrosis after injury. Front. Pharmacol. 2015, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.D.; Jeong, J.Y.; Jung, E.Y.; Yang, H.S.; Lim, H.T.; Joo, S.T. The influence of fiber size distribution of type IIB on carcass traits and meat quality in pigs. Meat. Sci. 2013, 94, 267–273. [Google Scholar] [CrossRef]
- Talbot, J.; Maves, L. Skeletal muscle fiber type, using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle, molecular regulation of myogenesis. Cold Spring Harb Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chen, W.; Wang, L.; You, W.; Wang, Y.; Wang, Y.; Zhao, J.; Shan, T. UCP1 Knockin Induces Lipid Dynamics and Transcriptional Programs in the Skeletal Muscles of Pigs. Front. Cell Dev. Biol. 2021, 9, 808095. [Google Scholar] [CrossRef]
- Zhao, P.; Hoffman, E.P. Embryonic myogenesis pathways in muscle regeneration. Dev. Dyn. 2004, 229, 380–392. [Google Scholar] [CrossRef]
- Davoli, R.; Braglia, S.; Russo, V.; Varona, L.; te Pas, M.F. Expression profiling of functional genes in prenatal skeletal muscle tissue in Duroc and Pietrain pigs. J. Anim. Breed. Genet. 2011, 128, 15–27. [Google Scholar] [CrossRef]
- Jin, Z.; Choe, H.M.; Lv, S.; Chang, S.; Yin, X. Esophageal striated muscle hypertrophy and muscle fiber type transformation in MSTN knockout pigs. Transgenic. Res. 2022, 31, 341–349. [Google Scholar] [CrossRef]
- Hennebry, A.; Oldham, J.; Shavlakadze, T.; Grounds, M.D.; Sheard, P.; Fiorotto, M.L.; Falconer, S.; Smith, H.K.; Berry, C.; Jeanplong, F.; et al. IGF1 stimulates greater muscle hypertrophy in the absence of myostatin in male mice. J. Endocrinol. 2017, 234, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Lautherbach, N.; Goncalves, D.A.P.; Silveira, W.A.; Paula-Gomes, S.; Valentim, R.R.; Zanon, N.M.; Pereira, M.G.; Miyabara, E.H.; Navegantes, L.C.C.; Kettelhut, I.C. Urocortin 2 promotes hypertrophy and enhances skeletal muscle function through cAMP and insulin/IGF-1 signaling pathways. Mol. Metab. 2022, 60, 101492. [Google Scholar] [CrossRef]
- Liu, H.; Xi, Y.; Liu, G.; Zhao, Y.; Li, J.; Lei, M. Comparative transcriptomic analysis of skeletal muscle tissue during prenatal stages in Tongcheng and Yorkshire pig using RNA-seq. Funct. Integr. Genom. 2018, 18, 195–209. [Google Scholar] [CrossRef]
- Magri, M.S.; Voronov, D.; Randelovic, J.; Cuomo, C.; Gomez-Skarmeta, J.L.; Arnone, M.I. ATAC-Seq for Assaying Chromatin Accessibility Protocol Using Echinoderm Embryos. Methods Mol. Biol. 2021, 2219, 253–265. [Google Scholar]
- Ming, H.; Sun, J.; Pasquariello, R.; Gatenby, L.; Herrick, J.R.; Yuan, Y.; Pinto, C.R.; Bondioli, K.R.; Krisher, R.L.; Jiang, Z. The landscape of accessible chromatin in bovine oocytes and early embryos. Epigenetics 2021, 16, 300–312. [Google Scholar] [CrossRef]
- Yue, J.; Hou, X.; Liu, X.; Wang, L.; Gao, H.; Zhao, F.; Shi, L.; Shi, L.; Yan, H.; Deng, T.; et al. The landscape of chromatin accessibility in skeletal muscle during embryonic development in pigs. J. Anim. Sci. Biotechnol. 2021, 12, 56. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Mohammadabadi, M.; Bordbar, F.; Jensen, J.; Du, M.; Guo, W. Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals. Animals 2021, 11, 835. [Google Scholar] [CrossRef] [PubMed]
- Poghosyan, T.; Gaujoux, S.; Vanneaux, V.; Bruneval, P.; Domet, T.; Lecourt, S.; Jarraya, M.; Sfeir, R.; Larghero, J.; Cattan, P. In vitro development and characterization of a tissue-engineered conduit resembling esophageal wall using human and pig skeletal myoblast, oral epithelial cells, and biologic scaffolds. Tissue Eng. Part A 2013, 19, 2242–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kacperczyk, A.; Jagla, T.; Daczewska, M. Pax-3 and Pax-7 label muscle progenitor cells during myotomal myogenesis in Coregonus lavaretus (Teleostei, Coregonidae). Anat. Histol. Embryol. 2009, 38, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Han, S.H.; Kim, S.G.; Kim, S.Y.; Kim, H.A.; Yoo, J.H.; Shin, M.C.; Yang, B.C.; Lee, J.H.; Cho, I.C. The MYH3 gene variant (g.-1805_-1810, ACGT) affects endophenotypes of myoglobin oxidation-reduction states in porcine muscles. Anim. Genet. 2022, 53, 532–533. [Google Scholar] [CrossRef]
- Cho, I.C.; Park, H.B.; Ahn, J.S.; Han, S.H.; Lee, J.B.; Lim, H.T.; Yoo, C.K.; Jung, E.J.; Kim, D.H.; Sun, W.S.; et al. A functional regulatory variant of MYH3 influences muscle fiber-type composition and intramuscular fat content in pigs. PLoS Genet. 2019, 15, e1008279. [Google Scholar] [CrossRef] [Green Version]
- Han, S.Z.; Gao, K.; Chang, S.Y.; Choe, H.M.; Paek, H.J.; Quan, B.H.; Liu, X.Y.; Yang, L.H.; Lv, S.T.; Yin, X.J.; et al. miR-455-3p Is Negatively Regulated by Myostatin in Skeletal Muscle and Promotes Myoblast Differentiation. J. Agric. Food Chem. 2022, 70, 10121–10133. [Google Scholar] [CrossRef]
- Howlett, K.F.; McGee, S.L. Epigenetic regulation of skeletal muscle metabolism. Clin. Sci. 2016, 130, 1051–1063. [Google Scholar] [CrossRef]
- Laker, R.C.; Ryall, J.G. DNA Methylation in Skeletal Muscle Stem Cell Specification, Proliferation, and Differentiation. Stem Cells Int. 2016, 2016, 5725927. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.M.; Zhou, J.; Quan, R.; Hong, L.J.; Li, Z.C.; Zheng, E.Q.; Liu, W.; Wu, Z.F.; Cai, G.Y.; Gu, T. Histone H3K27me3 in the regulation of skeletal muscle development. Yi Chuan 2019, 41, 285–292. [Google Scholar]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef] [Green Version]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [Green Version]
- Tachiwana, H.; Osakabe, A.; Shiga, T.; Miya, Y.; Kimura, H.; Kagawa, W.; Kurumizaka, H. Structures of human nucleosomes containing major histone H3 variants. Acta. Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 6, 578–583. [Google Scholar] [CrossRef]
- Tsunaka, Y.; Furukawa, A.; Nishimura, Y. Histone tail network and modulation in a nucleosome. Curr. Opin. Struct. Biol. 2022, 75, 102436. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Kissane, S.; Dhandapani, V.; Orsini, L. Protocol for assay of transposase accessible chromatin sequencing in non-model species. STAR Protoc. 2021, 2, 100341. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq, A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Ma, Z.; Tang, Z.; Yu, L.; Liu, S.; Huang, T.; Wang, P.; Wu, T.; Song, Z.; Zhang, H.; et al. Integrative ATAC-seq and RNA-seq Analysis of the Longissimus Muscle of Luchuan and Duroc Pigs. Front. Nutr. 2021, 8, 742672. [Google Scholar] [CrossRef]
- Salavati, M.; Woolley, S.A.; Cortes Araya, Y.; Halstead, M.M.; Stenhouse, C.; Johnsson, M.; Ashworth, C.J.; Archibald, A.L.; Donadeu, F.X.; Hassan, M.A.; et al. Profiling of open chromatin in developing pig (Sus scrofa) muscle to identify regulatory regions. G3 Bethesda 2022, 12, jkab424. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Wu, W.; Hou, L.; Chen, H.; Zuo, B.; Xiong, Y.; Yang, J. Skeletal Muscle-Specific Overexpression of PGC-1alpha Induces Fiber-Type Conversion through Enhanced Mitochondrial Respiration and Fatty Acid Oxidation in Mice and Pigs. Int. J. Biol. Sci. 2017, 13, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Grefte, S.; Kuijpers-Jagtman, A.M.; Torensma, R.; Von den Hoff, J.W. Skeletal muscle development and regeneration. Stem Cells Dev. 2007, 16, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Chen, H.; Liu, W.; Liu, H.; Gong, J.; Wang, H.; Guo, A.Y. AnimalTFDB, a comprehensive animal transcription factor database. Nucleic Acids Res. 2012, 40, D144–D149. [Google Scholar] [CrossRef] [PubMed]
- Haluskova, J. Epigenetic studies in human diseases. Folia Biol. Praha 2010, 56, 83–96. [Google Scholar] [PubMed]
- Blewitt, M.; Whitelaw, E. The use of mouse models to study epigenetics. Cold. Spring Harb Perspect. Biol. 2013, 5, a017939. [Google Scholar] [CrossRef] [PubMed]
- Schiele, M.A.; Domschke, K. Epigenetics at the crossroads between genes, environment and resilience in anxiety disorders. Genes Brain Behav. 2018, 17, e12423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Dorantes, M.T.; Diaz-Lopez, Y.E.; Gutierrez-Aguilar, R. Environment and Gene Association With Obesity and Their Impact on Neurodegenerative and Neurodevelopmental Diseases. Front. Neurosci. 2020, 14, 863. [Google Scholar] [CrossRef]
- Dehghani, H. Regulation of Chromatin Organization in Cell Stemness. The Emerging Role of Long Non-coding RNAs. Stem Cell Rev. Rep. 2021, 17, 2042–2053. [Google Scholar] [CrossRef]
- Wang, P.; Yang, W.; Zhao, S.; Nashun, B. Regulation of chromatin structure and function, insights into the histone chaperone FACT. Cell Cycle 2021, 20, 465–479. [Google Scholar] [CrossRef]
- White, R.B.; Bierinx, A.S.; Gnocchi, V.F.; Zammit, P.S. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 2010, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yan, J.; Fan, X.; Chen, J.; Wang, Z.; Liu, X.; Yi, G.; Liu, Y.; Niu, Y.; Zhang, L.; et al. The genome variation and developmental transcriptome maps reveal genetic differentiation of skeletal muscle in pigs. PLoS Genet. 2021, 17, e1009910. [Google Scholar] [CrossRef]
- Stefani, C.; Miricescu, D.; Stanescu, S., II; Nica, R.I.; Greabu, M.; Totan, A.R.; Jinga, M. Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis, Where Are We Now? Int. J. Mol. Sci. 2021, 22, 10260. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, L.; Chen, H.; Ye, W.; Dong, S.; Zheng, X.; Wang, Y. ATAC-Seq Reveals the Landscape of Open Chromatin and cis-Regulatory Elements in the Phytophthora sojae Genome. Mol. Plant Microbe Interact. 2022, 35, 301–310. [Google Scholar] [CrossRef]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef]
- Chen, K.; Gao, P.; Li, Z.; Dai, A.; Yang, M.; Chen, S.; Su, J.; Deng, Z.; Li, L. Forkhead Box O Signaling Pathway in Skeletal Muscle Atrophy. Am. J. Pathol. 2022, 192, 1648–1657. [Google Scholar] [CrossRef]
- Girardi, F.; Le Grand, F. Wnt Signaling in Skeletal Muscle Development and Regeneration. Prog. Mol. Biol. Transl. Sci. 2018, 153, 157–179. [Google Scholar]
- Cisternas, P.; Henriquez, J.P.; Brandan, E.; Inestrosa, N.C. Wnt signaling in skeletal muscle dynamics, myogenesis, neuromuscular synapse and fibrosis. Mol. Neurobiol. 2014, 49, 574–589. [Google Scholar] [CrossRef]
- Yu, C.; Li, Y.; Zhang, B.; Lin, M.; Li, J.; Zhang, L.; Wang, T.; Gao, F.; Zhou, G. Suppression of mTOR Signaling Pathways in Skeletal Muscle of Finishing Pigs by Increasing the Ratios of Ether Extract and Neutral Detergent Fiber at the Expense of Starch in Iso-energetic Diets. J. Agric. Food Chem. 2016, 64, 1557–1564. [Google Scholar] [CrossRef]
- Ropka-Molik, K.; Bereta, A.; Zukowski, K.; Piorkowska, K.; Gurgul, A.; Zak, G. Transcriptomic gene profiling of porcine muscle tissue depending on histological properties. Anim. Sci. J. 2017, 88, 1178–1188. [Google Scholar] [CrossRef]
- Hou, Y.; Su, L.; Su, R.; Luo, Y.; Wang, B.; Yao, D.; Zhao, L.; Jin, Y. Effect of feeding regimen on meat quality, MyHC isoforms, AMPK, and PGC-1alpha genes expression in the biceps femoris muscle of Mongolia sheep. Food Sci. Nutr. 2020, 8, 2262–2270. [Google Scholar] [CrossRef] [Green Version]
- Kfoury, A.; Armaro, M.; Collodet, C.; Sordet-Dessimoz, J.; Giner, M.P.; Christen, S.; Moco, S.; Leleu, M.; de Leval, L.; Koch, U.; et al. AMPK promotes survival of c-Myc-positive melanoma cells by suppressing oxidative stress. EMBO J. 2018, 37, e97673. [Google Scholar] [CrossRef]
- Christoffolete, M.A.; Silva, W.J.; Ramos, G.V.; Bento, M.R.; Costa, M.O.; Ribeiro, M.O.; Okamoto, M.M.; Lohmann, T.H.; Machado, U.F.; Musaro, A.; et al. Muscle IGF-1-induced skeletal muscle hypertrophy evokes higher insulin sensitivity and carbohydrate use as preferential energy substrate. Biomed. Res. Int. 2015, 2015, 282984. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Tong, H.; Zhang, Z.; Shao, S.; Liu, D.; Li, S.; Yan, Y. Transcription factor EGR1 promotes differentiation of bovine skeletal muscle satellite cells by regulating MyoG gene expression. J. Cell Physiol. 2018, 233, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Marmol-Sanchez, E.; Quintanilla, R.; Jordana, J.; Amills, M. An association analysis for 14 candidate genes mapping to meat quality quantitative trait loci in a Duroc pig population reveals that the ATP1A2 genotype is highly associated with muscle electric conductivity. Anim. Genet. 2020, 51, 95–100. [Google Scholar] [CrossRef]
- Deng, Y.; Bartosovic, M.; Ma, S.; Zhang, D.; Kukanja, P.; Xiao, Y.; Su, G.; Liu, Y.; Qin, X.; Rosoklija, G.B.; et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 2022, 609, 375–383. [Google Scholar] [CrossRef]
- Baek, S.; Lee, I. Single-cell ATAC sequencing analysis, From data preprocessing to hypothesis generation. Comput. Struct. Biotechnol. J. 2020, 18, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Bintu, L.; Yong, J.; Antebi, Y.E.; McCue, K.; Kazuki, Y.; Uno, N.; Oshimura, M.; Elowitz, M.B. Dynamics of epigenetic regulation at the single-cell level. Science 2016, 351, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Bo, Y.Y.; Liang, L.D.; Hua, Y.J.; Zhao, Z.; Yao, M.S.; Shan, L.B.; Liang, C.Z. High-purity DNA extraction from animal tissue using picking in the TRIzol-based method. Biotechniques 2021, 70, 186–190. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, L.; Si, J.; Yue, J.; Zhao, M.; Qi, W.; Zhu, S.; Mo, J.; Wang, L.; Lan, G.; Liang, J. The Landscape of Accessible Chromatin and Developmental Transcriptome Maps Reveal a Genetic Mechanism of Skeletal Muscle Development in Pigs. Int. J. Mol. Sci. 2023, 24, 6413. https://doi.org/10.3390/ijms24076413
Feng L, Si J, Yue J, Zhao M, Qi W, Zhu S, Mo J, Wang L, Lan G, Liang J. The Landscape of Accessible Chromatin and Developmental Transcriptome Maps Reveal a Genetic Mechanism of Skeletal Muscle Development in Pigs. International Journal of Molecular Sciences. 2023; 24(7):6413. https://doi.org/10.3390/ijms24076413
Chicago/Turabian StyleFeng, Lingli, Jinglei Si, Jingwei Yue, Mingwei Zhao, Wenjing Qi, Siran Zhu, Jiayuan Mo, Lixian Wang, Ganqiu Lan, and Jing Liang. 2023. "The Landscape of Accessible Chromatin and Developmental Transcriptome Maps Reveal a Genetic Mechanism of Skeletal Muscle Development in Pigs" International Journal of Molecular Sciences 24, no. 7: 6413. https://doi.org/10.3390/ijms24076413