
Novel Cannabinoid Receptor 2 (CB2) Low Lipophilicity Agonists Produce Distinct cAMP and Arrestin Signalling Kinetics without Bias
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Compound Properties and Binding Affinities
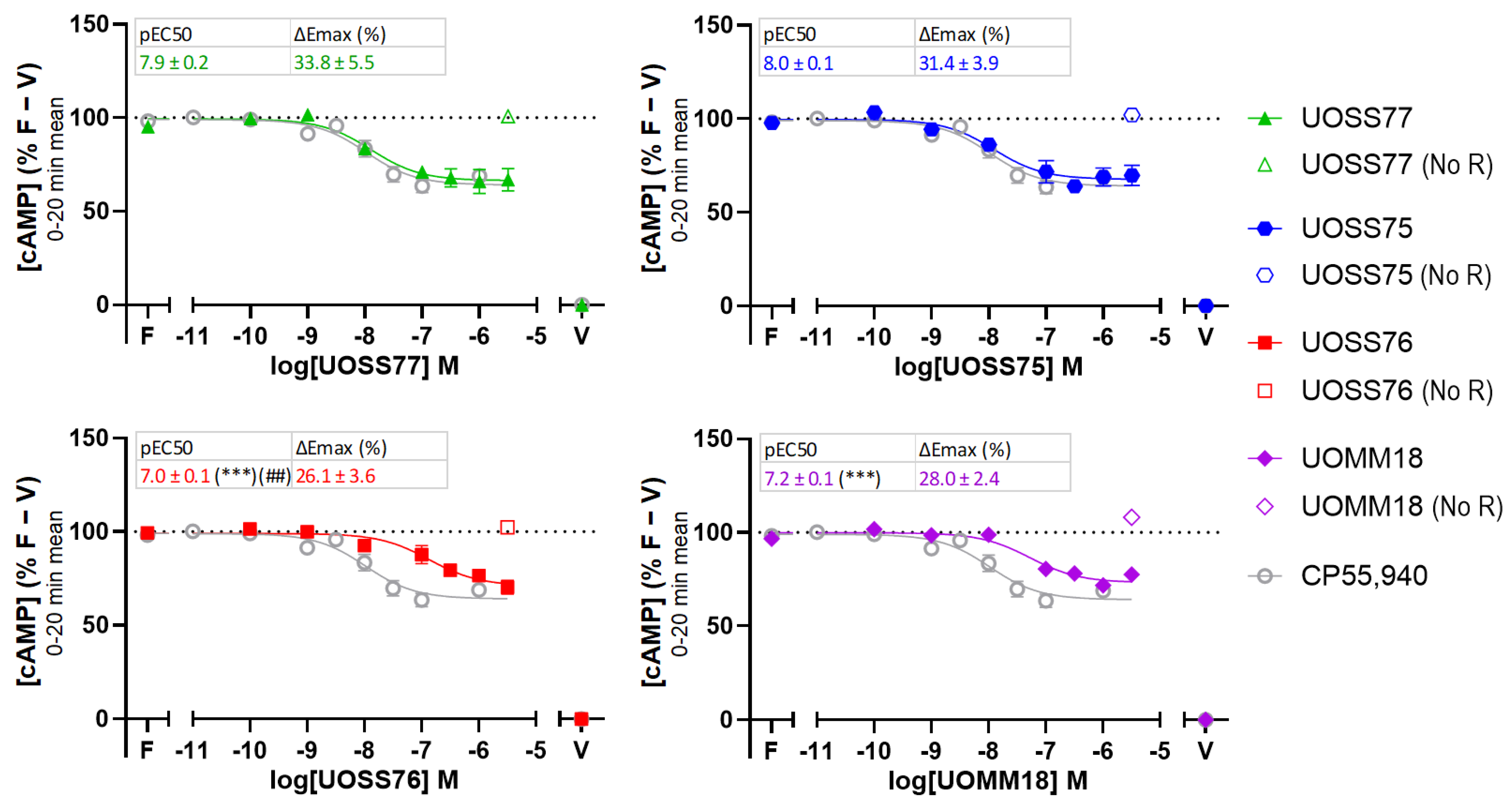
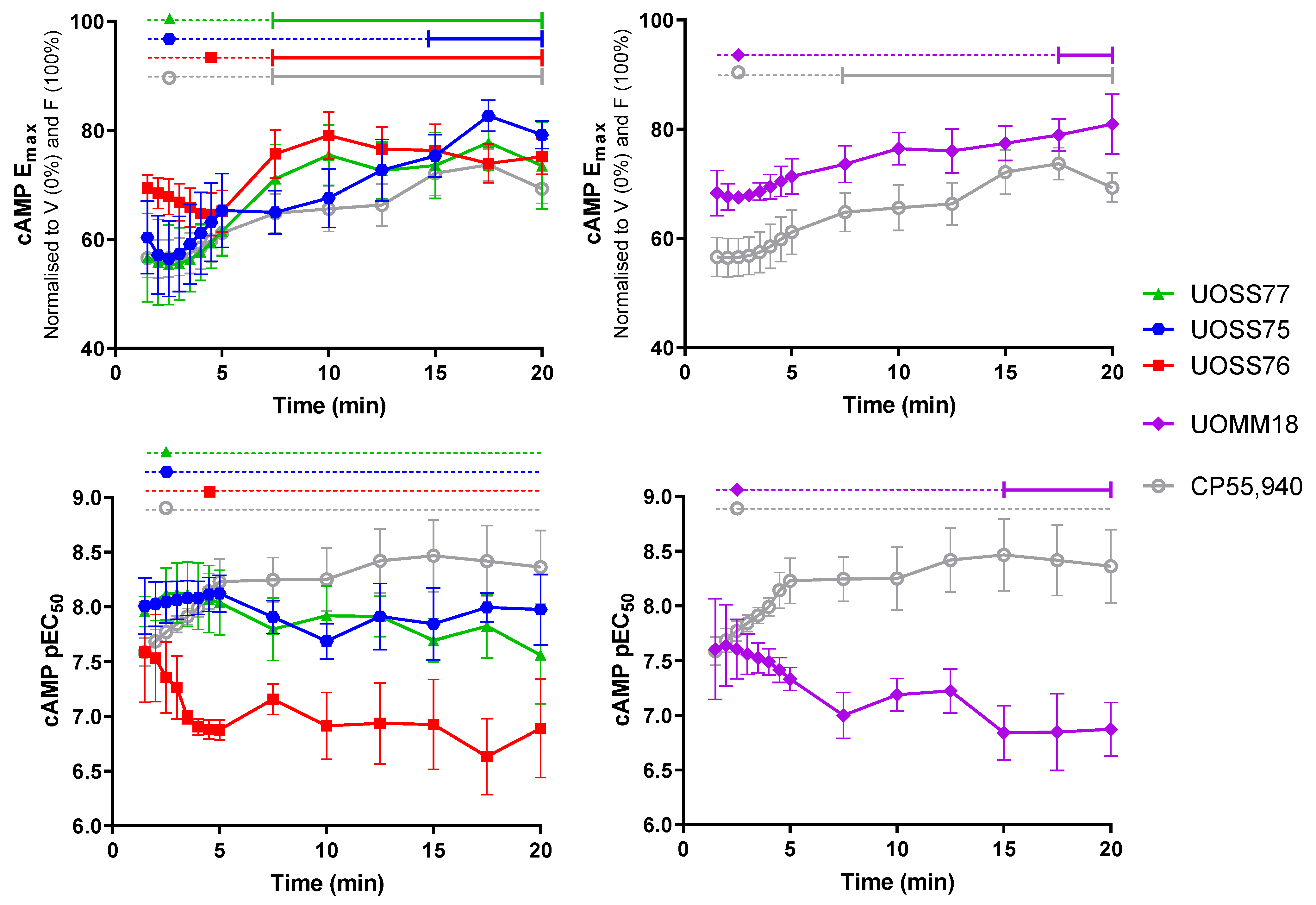
2.2. cAMP Signalling
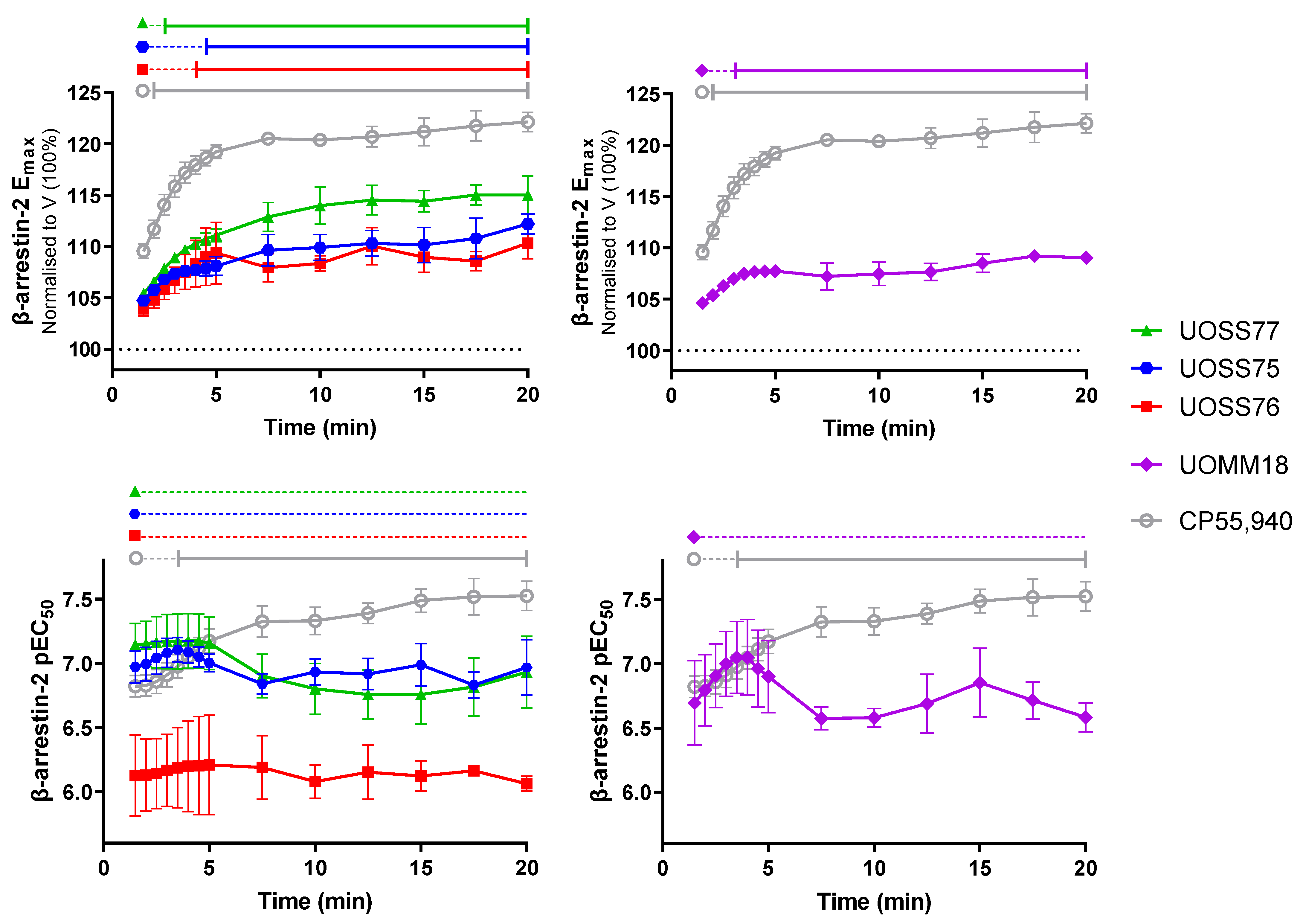
2.3. β-Arrestin-2 Recruitment
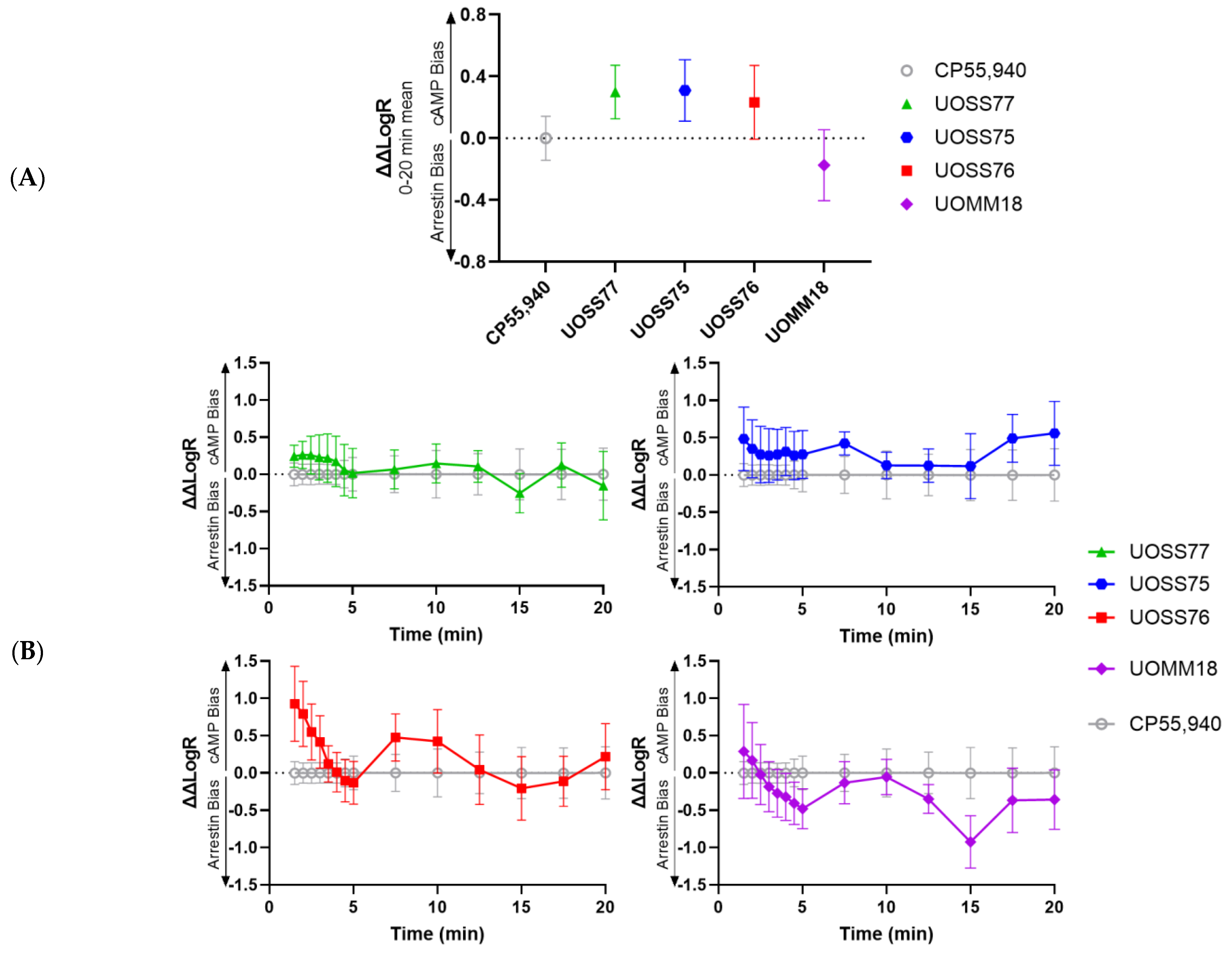
2.4. Bias Analysis
3. Discussion
4. Materials and Methods
4.1. Compound Synthesis and Parameters
4.2. Competition Radioligand Binding Assay
4.3. cAMP and β-Arrestin-2 Assay Cell Plating and Transfection
4.4. cAMP and β-Arrestin-2 Assay Procedure and Analysis
4.5. Bias Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Turcotte, C.; Blanchet, M.-R.; Laviolette, M.; Flamand, N. The CB 2 Receptor and Its Role as a Regulator of Inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef]
- Whiting, Z.M.; Yin, J.; de la Harpe, S.M.; Vernall, A.J.; Grimsey, N.L. Developing the Cannabinoid Receptor 2 (CB2) Pharmacopoeia: Past, Present, and Future. Trends Pharmacol. Sci. 2022, 43, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xiao, P.; Sun, J. Structural Basis of Signaling of Cannabinoids Receptors: Paving a Way for Rational Drug Design in Controling Mutiple Neurological and Immune Diseases. Signal Transduct. Target. Ther. 2020, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Calapai, F.; Cardia, L.; Sorbara, E.E.; Navarra, M.; Gangemi, S.; Calapai, G.; Mannucci, C. Cannabinoids, Blood–Brain Barrier, and Brain Disposition. Pharmaceutics 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Saccomanni, G.; Adinolfi, B.; Benetti, V.; Ligresti, A.; Cascio, M.G.; Tuccinardi, T.; Lucchesi, V.; Martinelli, A.; Nieri, P.; et al. Rational Design, Synthesis, and Pharmacological Properties of New 1,8-Naphthyridin-2(1H)-on-3-Carboxamide Derivatives as Highly Selective Cannabinoid-2 Receptor Agonists. J. Med. Chem. 2009, 52, 3644–3651. [Google Scholar] [CrossRef]
- Odan, M.; Ishizuka, N.; Hiramatsu, Y.; Inagaki, M.; Hashizume, H.; Fujii, Y.; Mitsumori, S.; Morioka, Y.; Soga, M.; Deguchi, M.; et al. Discovery of S-777469: An Orally Available CB2 Agonist as an Antipruritic Agent. Bioorganic Med. Chem. Lett. 2012, 22, 2803–2806. [Google Scholar] [CrossRef]
- Oyagawa, C.R.M.; De Harpe, S.M.; Saroz, Y.; Glass, M.; Vernall, A.J.; Grimsey, N.L. Cannabinoid Receptor 2 Signalling Bias Elicited by 2,4,6-Trisubstituted 1,3,5-Triazines. Front. Pharmacol. 2018, 9, 1202. [Google Scholar] [CrossRef] [PubMed]
- Shionogi & Co., Ltd. Process for Producing 1-Substituted-2-Pyridone-3-Carboxylic Acid Derivative (Patent WO2008084671). 2008. Available online: https://web.archive.org/web/20230122064416/https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008084671&_cid=P21-LA1KFF-65044-1 (accessed on 22 January 2023).
- Haruna, T.; Soga, M.; Morioka, Y.; Hikita, I.; Imura, K.; Furue, Y.; Yamamoto, M.; Imura, C.; Ikeda, M.; Yamauchi, A.; et al. S-777469, a Novel Cannabinoid Type 2 Receptor Agonist, Suppresses Itch-Associated Scratching Behavior in Rodents through Inhibition of Itch Signal Transmission. Pharmacology 2015, 95, 95–103. [Google Scholar] [CrossRef]
- Shionogi Inc. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Efficacy and Safety Study of 2 Doses of S-777469 (400 Mg BID and 800 Mg BID) in Patients With Atopic Dermatitis. NCT00703573. 2009. Available online: https://web.archive.org/web/20210403093153/https://clinicaltrials.gov/ct2/show/NCT00703573 (accessed on 3 April 2021).
- Shionogi & Co. 1st Quarter of Fiscal 2009 Conference Call. Available online: https://web.archive.org/web/20220627232021/https://www.shionogi.com/content/dam/shionogi/global/investors/pdf/e_p090803.pdf (accessed on 27 June 2022).
- Odan, M.; Ishizuka, N.; Hiramatsu, Y.; Inagaki, M.; Hashizume, H.; Fujii, Y.; Mitsumori, S.; Morioka, Y.; Soga, M.; Deguchi, M.; et al. Discovery of S-444823, a Potent CB1/CB2 Dual Agonist as an Antipruritic Agent. Bioorganic Med. Chem. Lett. 2012, 22, 2898–2901. [Google Scholar] [CrossRef]
- Odan, M.; Ishizuka, N.; Hiramatsu, Y.; Inagaki, M.; Hashizume, H.; Fujii, Y.; Mitsumori, S.; Morioka, Y.; Soga, M.; Deguchi, M.; et al. CB 1/2 Dual Agonists with 3-Carbamoyl 2-Pyridone Derivatives as Antipruritics: Reduction of CNS Side Effects by Introducing Polar Functional Groups. Bioorganic Med. Chem. Lett. 2012, 22, 2894–2897. [Google Scholar] [CrossRef]
- Lucchesi, V.; Hurst, D.P.; Shore, D.M.; Bertini, S.; Ehrmann, B.M.; Allara, M.; Lawrence, L.; Ligresti, A.; Minutolo, F.; Saccomanni, G.; et al. CB2-Selective Cannabinoid Receptor Ligands: Synthesis, Pharmacological Evaluation, and Molecular Modeling Investigation of 1,8-Naphthyridin-2(1H)-one-3-Carboxamides. J. Med. Chem. 2014, 57, 8777–8791. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Laezza, C.; Bertini, S.; Marasco, D.; Tuccinardi, T.; Bifulco, M.; Manera, C. Immunomodulatory Properties of 1,2-Dihydro-4-Hydroxy-2-Oxo-1,8-Naphthyridine-3-Carboxamide Derivative VL15. Biochimie 2017, 135, 173–180. [Google Scholar] [CrossRef]
- Cooper, A.G.; Oyagawa, C.R.M.; Manning, J.J.; Singh, S.; Hook, S.; Grimsey, N.L.; Glass, M.; Tyndall, J.D.A.; Vernall, A.J. Development of Selective, Fluorescent Cannabinoid Type 2 Receptor Ligands Based on a 1,8-Naphthyridin-2-(1: H)-One-3-Carboxamide Scaffold. Medchemcomm 2018, 9, 2055–2067. [Google Scholar] [CrossRef]
- Van Der Westhuizen, E.T.; Breton, B.; Christopoulos, A.; Bouvier, M. Quantification of Ligand Bias for Clinically Relevant B2-Adrenergic Receptor Ligands: Implications for Drug Taxonomy S. Mol. Pharmacol. 2014, 85, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Black, J.; Leff, P. Operational Models of Pharmacological Agonism. Biol. Sci. 1983, 220, 141–162. [Google Scholar]
- Ibsen, M.S.; Finlay, D.B.; Patel, M.; Javitch, J.A.; Glass, M.; Grimsey, N.L. Cannabinoid CB1 and CB2 Receptor-Mediated Arrestin Translocation: Species, Subtype, and Agonist-Dependence. Front. Pharmacol. 2019, 10, 350. [Google Scholar] [CrossRef]
- Herenbrink, C.K.; Sykes, D.A.; Donthamsetti, P.; Canals, M.; Coudrat, T.; Shonberg, J.; Scammells, P.J.; Capuano, B.; Sexton, P.M.; Charlton, S.J.; et al. The Role of Kinetic Context in Apparent Biased Agonism at GPCRs. Nat. Commun. 2016, 7, 10842. [Google Scholar] [CrossRef]
- Bologna, Z.; Teoh, J.P.; Bayoumi, A.S.; Tang, Y.; Kim, I.M. Biased g Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017, 25, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.B.; Cawston, E.E.; Grimsey, N.L.; Hunter, M.R.; Vemuri, V.K.; Makriyannis, A.; Glass, M. Gαs Signalling of the CB1 Receptor and the Influence of Receptor Number. Br. J. Pharmacol. 2017, 174, 2545–2562. [Google Scholar] [CrossRef]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low Intrinsic Efficacy for G Protein Activation Can Explain the Improved Side Effect Profiles of New Opioid Agonists. Sci. Signal. 2020, 13, eaaz3140. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E. V Biased GPCR Signaling: Possible Mechanisms and Inherent Limitations. Pharmacol Ther. 2020, 211, 107540. [Google Scholar] [CrossRef]
- Grundmann, M.; Kostenis, E. Temporal Bias: Time-Encoded Dynamic GPCR Signaling. Trends Pharmacol. Sci. 2017, 38, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Truong, T.T.; Merlin, J.; Sexton, P.M.; Wootten, D. Implications of Ligand-Receptor Binding Kinetics on GLP-1R Signalling. Biochem. Pharmacol. 2022, 199, 114985. [Google Scholar] [CrossRef] [PubMed]
- Soethoudt, M.; Hoorens, M.W.H.; Doelman, W.; Martella, A.; van der Stelt, M.; Heitman, L.H. Structure-Kinetic Relationship Studies of Cannabinoid CB2 Receptor Agonists Reveal Substituent-Specific Lipophilic Effects on Residence Time. Biochem. Pharmacol. 2018, 152, 129–142. [Google Scholar] [CrossRef]
- Vauquelin, G. Cell Membranes… and How Long Drugs May Exert Beneficial Pharmacological Activity in Vivo. Br. J. Clin. Pharmacol. 2016, 82, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.A.; Stoddart, L.A.; Kilpatrick, L.E.; Hill, S.J. Binding Kinetics of Ligands Acting at GPCRs. Mol. Cell. Endocrinol. 2019, 485, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-Coupled Receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef] [PubMed]
- Casajuana-Martin, N.; Navarro, G.; Gonzalez, A.; Llinas Del Torrent, C.; Gómez-Autet, M.; Quintana García, A.; Franco, R.; Pardo, L. A Single Point Mutation Blocks the Entrance of Ligands to the Cannabinoid CB2 Receptor via the Lipid Bilayer. J. Chem. Inf. Model. 2022, 62, 5771–5779. [Google Scholar] [CrossRef]
- Szlenk, C.T.; Gc, J.B.; Natesan, S. Does the Lipid Bilayer Orchestrate Access and Binding of Ligands to Transmembrane Orthosteric/Allosteric Sites of G Protein-Coupled Receptors? Mol. Pharmacol. 2019, 96, 527–541. [Google Scholar] [CrossRef]
- Cawston, E.E.; Redmond, W.J.; Breen, C.M.; Grimsey, N.L.; Connor, M.; Glass, M. Real-Time Characterization of Cannabinoid Receptor 1 (CB 1) Allosteric Modulators Reveals Novel Mechanism of Action. Br. J. Pharmacol. 2013, 170, 893–907. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Tikhonova, I.G.; Hudson, B.D.; Smith, N.J.; Mohr, K.; Ulven, T.; Milligan, G.; Kenakin, T.; Kostenis, E. A Molecular Mechanism for Sequential Activation of a G Protein-Coupled Receptor. Cell Chem. Biol. 2016, 23, 392–403. [Google Scholar] [CrossRef]
- Saleh, N.; Hucke, O.; Kramer, G.; Schmidt, E.; Montel, F.; Lipinski, R.; Ferger, B.; Clark, T.; Hildebrand, P.W.; Tautermann, C.S. Multiple Binding Sites Contribute to the Mechanism of Mixed Agonistic and Positive Allosteric Modulators of the Cannabinoid CB1 Receptor. Angew. Chemie 2018, 130, 2610–2615. [Google Scholar] [CrossRef]
- Egyed, A.; Kiss, D.J.; Keserű, G.M. The Impact of the Secondary Binding Pocket on the Pharmacology of Class A GPCRs. Front. Pharmacol. 2022, 13, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.W.; Holst, B. Ago-Allosteric Modulation and Other Types of Allostery in Dimeric 7TM Receptors. J. Recept. Signal Transduct. 2006, 26, 107–128. [Google Scholar] [CrossRef]
- Franco, R.; Morales, P.; Navarro, G.; Jagerovic, N.; Reyes-Resina, I. The Binding Mode to Orthosteric Sites and/or Exosites Underlies the Therapeutic Potential of Drugs Targeting Cannabinoid CB2 Receptors. Front. Pharmacol. 2022, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Ferrisi, R.; Polini, B.; Mohamed, K.A.; Ricardi, C.; Lucarini, E.; Carpi, S.; Domenichini, F.; Stevenson, L.A.; Rapposelli, S.; et al. Design, Synthesis, and Biological Activity of New CB2 Receptor Ligands: From Orthosteric and Allosteric Modulators to Dualsteric/Bitopic Ligands. J. Med. Chem. 2022, 65, 9918–9938. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Roy, K.K.; Doerksen, R.J. Negative Allosteric Modulators of Cannabinoid Receptor 2: Protein Modeling, Binding Site Identification and Molecular Dynamics Simulations in the Presence of an Orthosteric Agonist. J. Biomol. Struct. Dyn. 2020, 38, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Jiang, C.; Wang, J.; Chen, C.J.; Hao, Y.; Zhao, G.; Feng, Z.; Xie, X.Q. In Silico Prediction and Validation of CB2 Allosteric Binding Sites to Aid the Design of Allosteric Modulators. Molecules 2022, 27, 453. [Google Scholar] [CrossRef]
- Atay, O.; Skotheim, J.M. Spatial and Temporal Signal Processing and Decision Making by MAPK Pathways. J. Cell Biol. 2017, 216, 317–330. [Google Scholar] [CrossRef]
- Copeland, R.A. The Drug–Target Residence Time Model: A 10-year Retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef]
- Guo, D.; Hilliger, J.M.; Ijzerman, A.P.; Heitman, L.H. Drug-Target Residence Time—A Case for G Protein-Coupled Receptors Dong. Med. Res. Rev. 2014, 34, 856–892. [Google Scholar] [CrossRef]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic Monoacylglycerol Lipase Blockade Causes Functional Antagonism of the Endocannabinoid System. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Sharma, R.; Tyndall, J.D.A.; Grimsey, N.L.; Vernall, A.J. Synthesis and Characterization of a Cannabinoid Type 2 Receptor Photoactivated Prodrug. ChemPhotoChem 2023, e202200291. [Google Scholar] [CrossRef]
- Motulsky, H.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting; GraphPad Software Inc.: San Diego, CA, USA, 2003; p. 351. [Google Scholar]
- Wootten, D.; Lindmark, H.; Kadmiel, M.; Willcockson, H.; Caron, K.M.; Barwell, J.; Drmota, T.; Poyner, D.R. Receptor Activity Modifying Proteins (RAMPs) Interact with the VPAC 2 Receptor and CRF1 Receptors and Modulate Their Function. Br. J. Pharmacol. 2013, 168, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.; Decamp, D.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; Sternweis, P.C. Use of a CAMP BRET Sensor to Characterize a Novel Regulation of CAMP by the Sphingosine 1-Phosphate/G13 Pathway. J. Biol. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef]
- Clayton, C.C.; Donthamsetti, P.; Lambert, N.A.; Javitch, J.A.; Neve, K.A. Mutation of Three Residues in the Third Intracellular Loop of the Dopamine D2 Receptor Creates an Internalization-Defective Receptor. J. Biol. Chem. 2014, 289, 33663–33675. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | Molecular Weight (Da) | TPSA (Å2) | cLogP (cLogD7.4) | CB2 pKd/pKi (±SEM) | CB1 pKd/pKi (±SEM) | CB2/CB1 Selectivity |
|---|---|---|---|---|---|---|---|
| UOSS77/S-777469 a |  | 414.5 | 86.7 | 3.13 (−0.25) | 6.73 (0.04) | ~4.80 (0.08) | ~80 |
| UOSS75 |  | 545.7 | 117.2 | 1.44 (1.44) | 6.83 (0.13) | ~4.82 (0.21) | ~100 |
| UOSS76 |  | 589.7 | 126.4 | 1.27 (1.27) | 6.09 (0.18) | ~4.53 (0.16) | ~40 |
| UOMM18 |  | 514.5 | 95.0 | −0.19 (−0.20) | 6.14 (0.09) | <4 | >126 |
| CP55,940 |  | 376.6 | 60.7 | 5.57 (5.57) | 8.58 (0.09) | 7.99 (0.12) | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, R.; Singh, S.; Whiting, Z.M.; Molitor, M.; Vernall, A.J.; Grimsey, N.L. Novel Cannabinoid Receptor 2 (CB2) Low Lipophilicity Agonists Produce Distinct cAMP and Arrestin Signalling Kinetics without Bias. Int. J. Mol. Sci. 2023, 24, 6406. https://doi.org/10.3390/ijms24076406
Sharma R, Singh S, Whiting ZM, Molitor M, Vernall AJ, Grimsey NL. Novel Cannabinoid Receptor 2 (CB2) Low Lipophilicity Agonists Produce Distinct cAMP and Arrestin Signalling Kinetics without Bias. International Journal of Molecular Sciences. 2023; 24(7):6406. https://doi.org/10.3390/ijms24076406
Chicago/Turabian StyleSharma, Raahul, Sameek Singh, Zak M. Whiting, Maximilian Molitor, Andrea J. Vernall, and Natasha L. Grimsey. 2023. "Novel Cannabinoid Receptor 2 (CB2) Low Lipophilicity Agonists Produce Distinct cAMP and Arrestin Signalling Kinetics without Bias" International Journal of Molecular Sciences 24, no. 7: 6406. https://doi.org/10.3390/ijms24076406