
Effect of Single-Residue Mutations on CTCF Binding to DNA: Insights from Molecular Dynamics Simulations

Abstract
:1. Introduction

2. Results
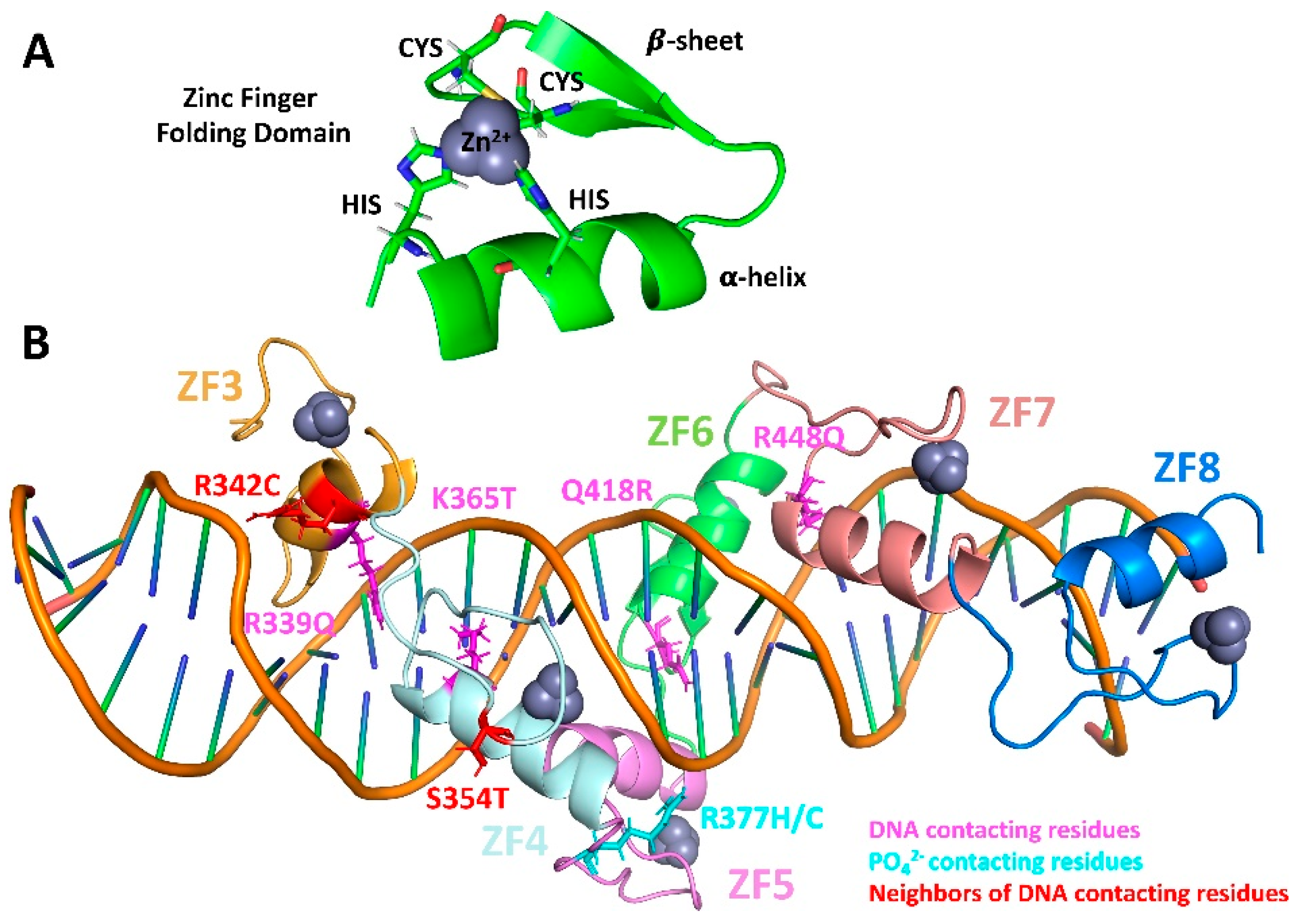
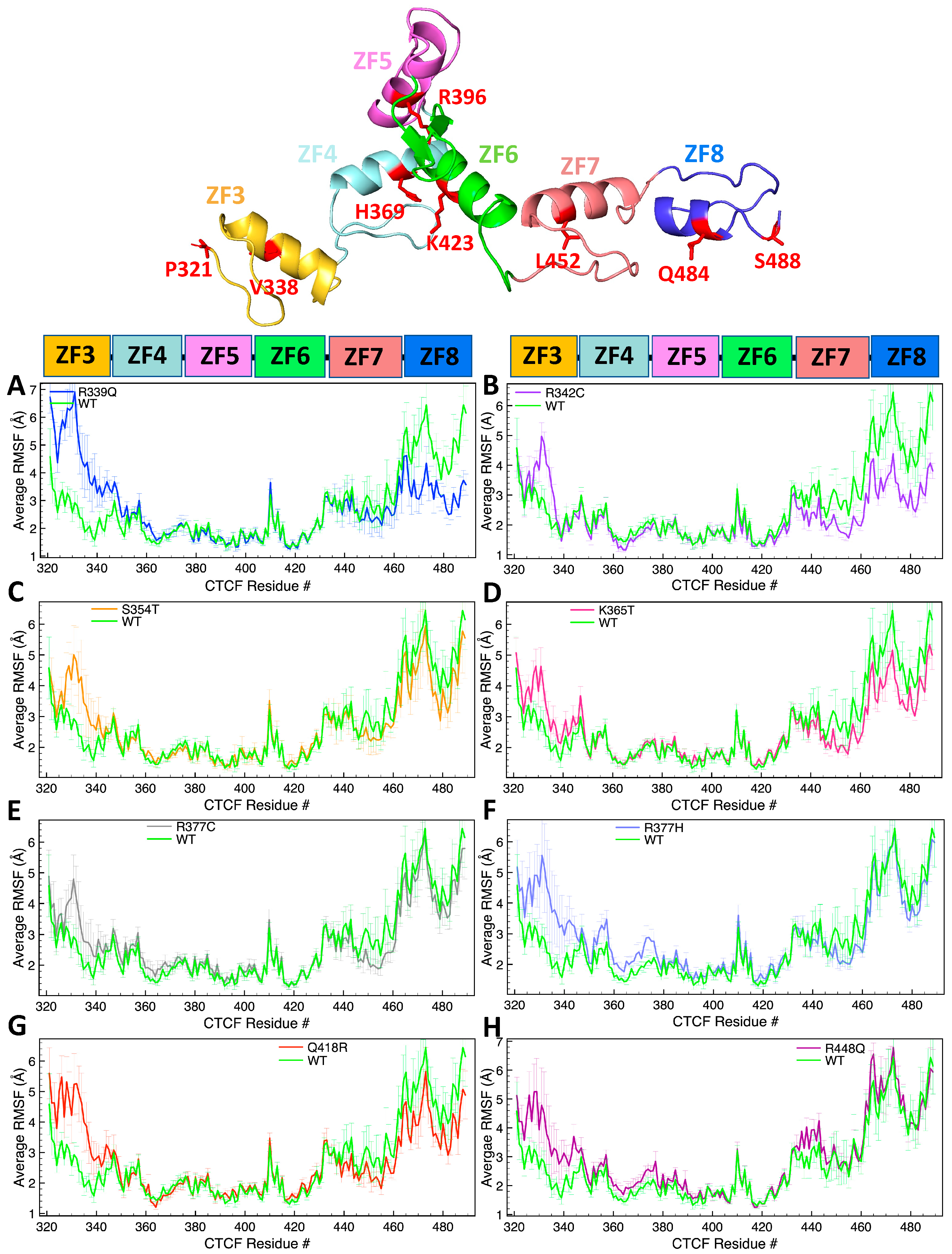
2.1. Single-Residue Mutations Affect Global CTCF Tertiary Structure and Flexibility
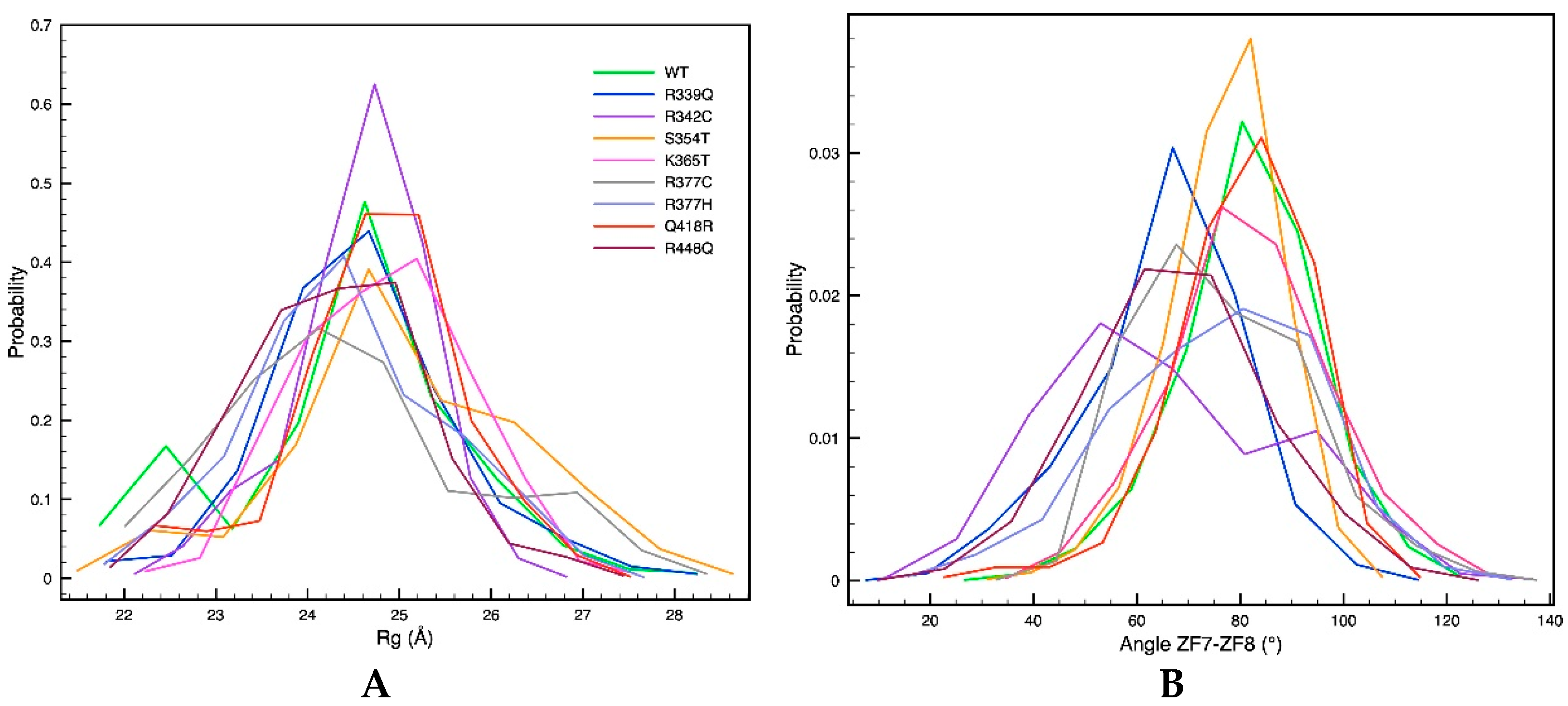
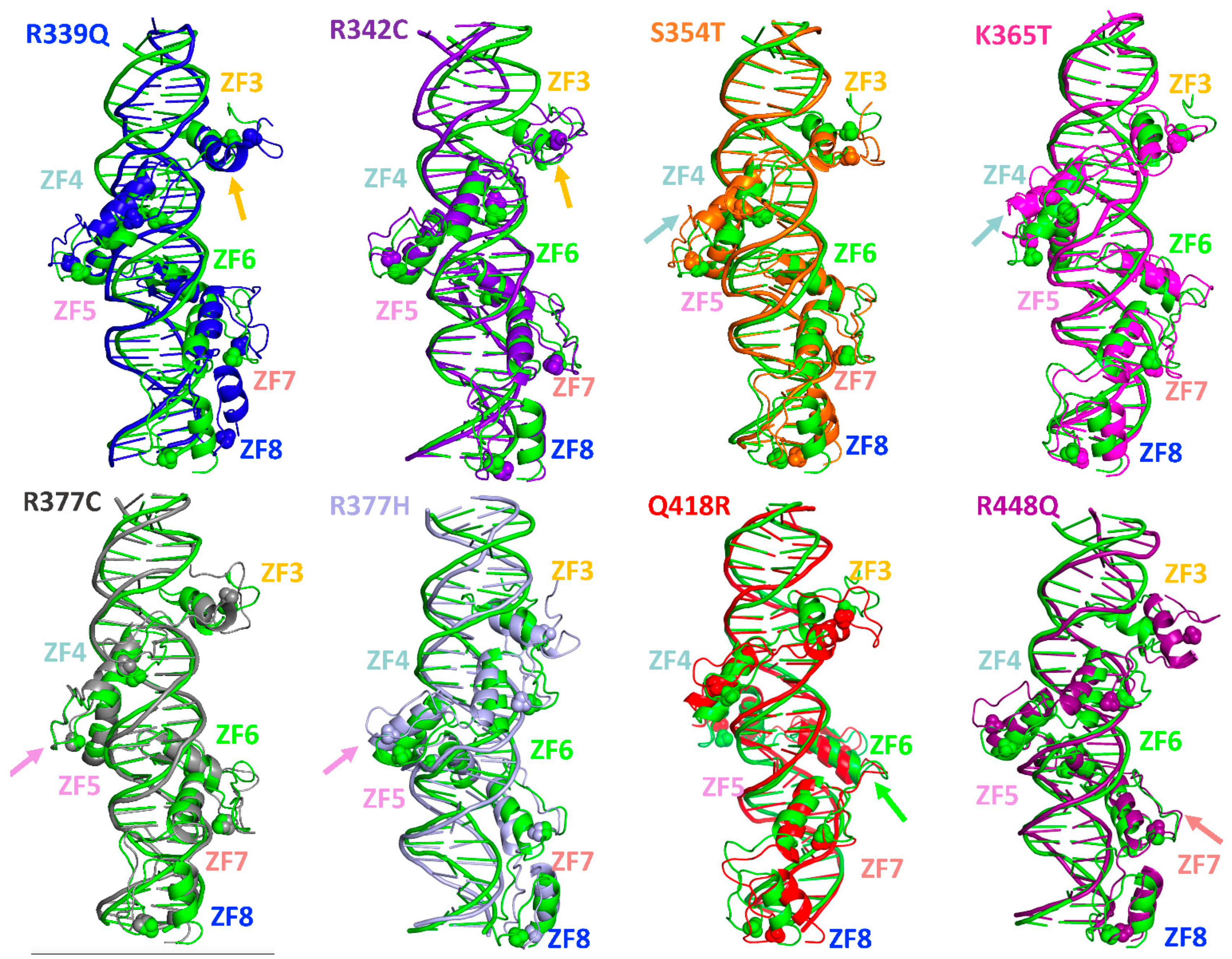
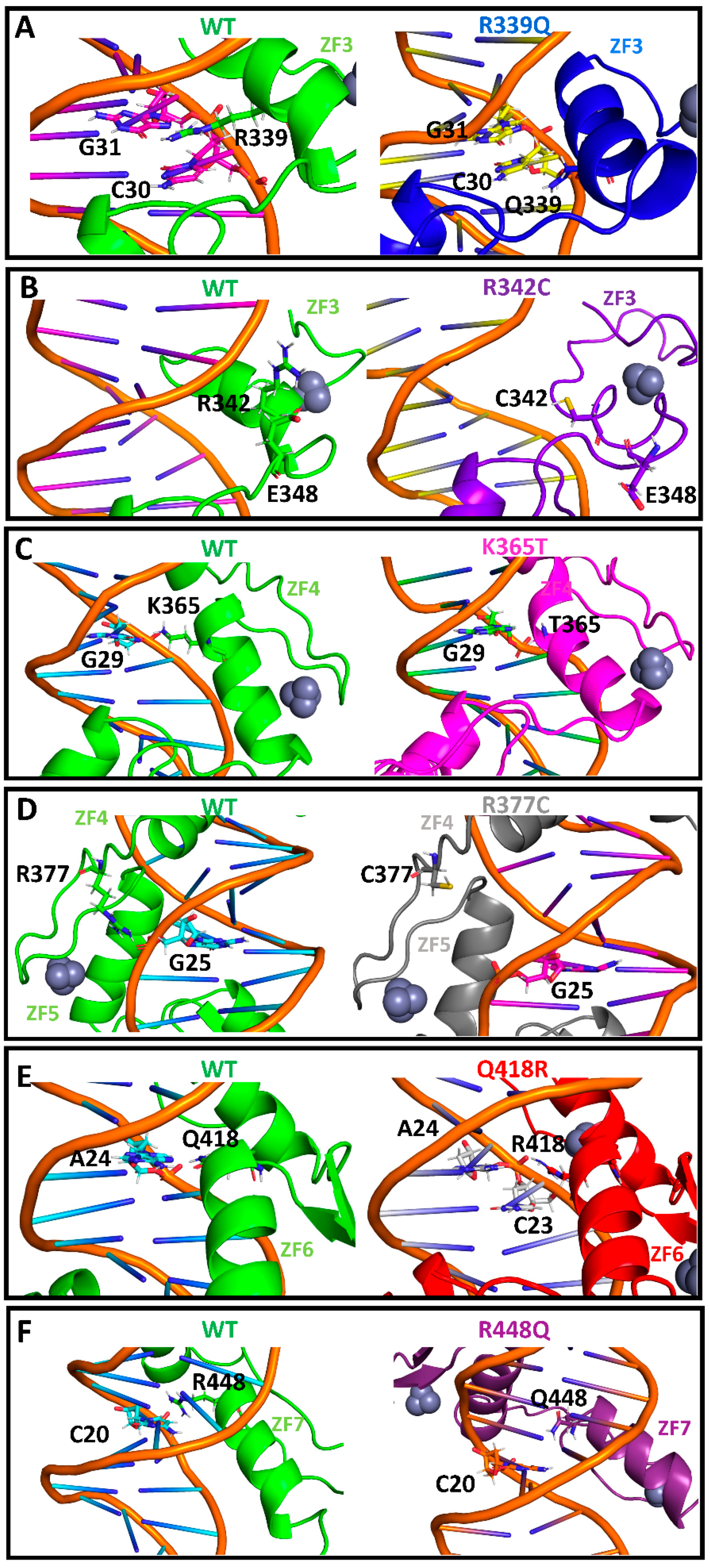
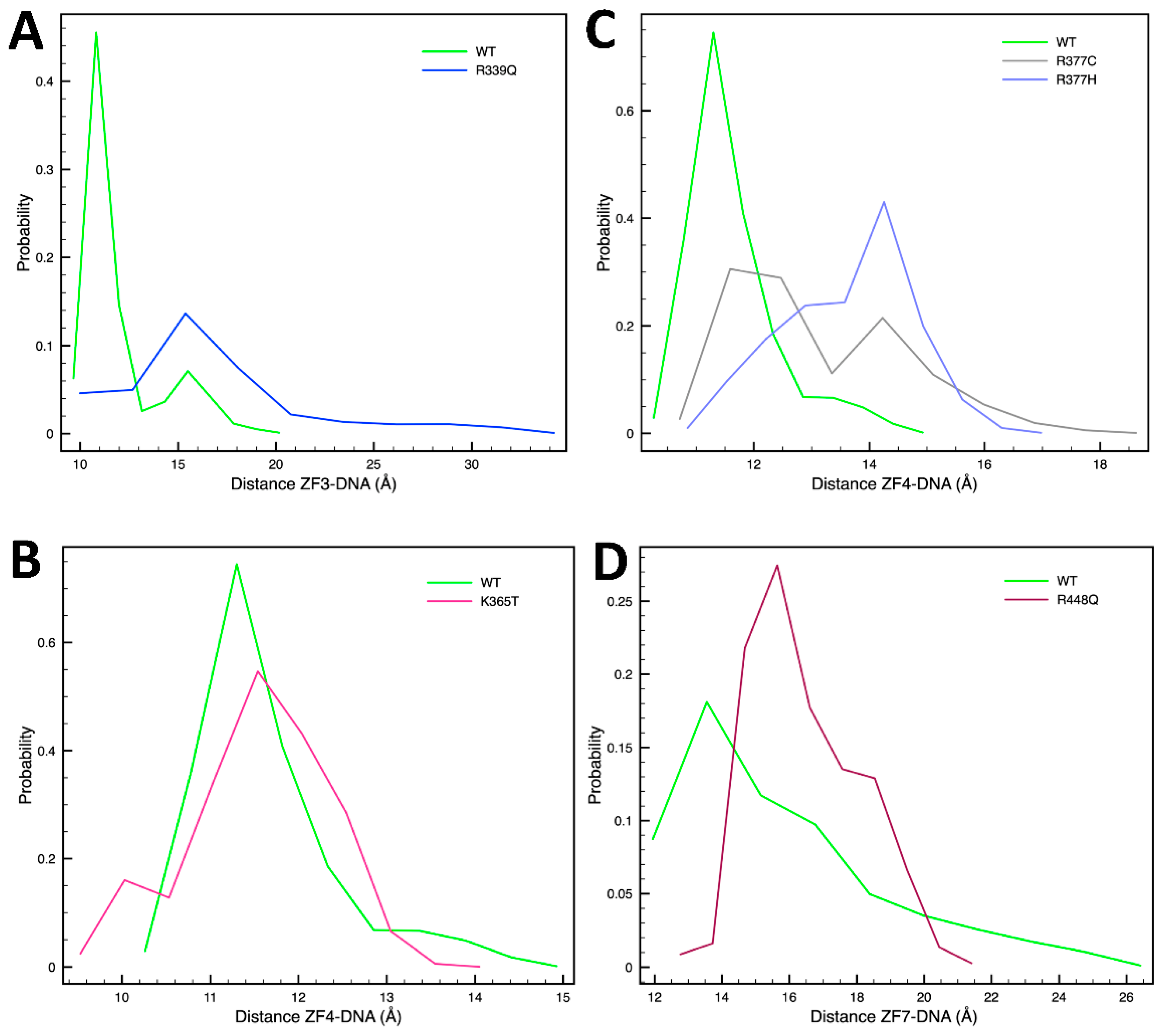
2.2. Single-Residue Mutations Affect the CTCF–DNA Complex Stability
3. Discussion
4. Materials and Methods
4.1. Initial Systems Models
4.2. Molecular Dynamics Simulations
4.3. Binding Free Energy Calculations
4.4. Structural Analysis Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CG | Conjugated Gradient |
| CTCF | CCCTC-binding factor |
| MD | Molecular Dynamics |
| MM-GBSA | Molecular Mechanics Generalized Born Surface Area |
| Rg | Radius of gyration |
| RMSD | Root Mean Squared Deviation |
| RMSF | Root Mean Squared Fluctuation |
| SD | Steepest Descent |
| TAD | Topologically Associating Domain |
| WT | Wildtype |
| ZF | Zinc Finger |
References
- Kim, S.; Yu, N.K.; Kaang, B.K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksimenko, O.G.; Fursenko, D.V.; Belova, E.V.; Georgiev, P.G. CTCF As an Example of DNA-Binding Transcription Factors Containing Clusters of C2H2-Type Zinc Fingers. Acta Nat. 2021, 13, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Filippova, G.; Qi, C.F.; Ulmer, J.; Moore, J.; Ward, M.; Hu, Y.; Loukinov, D.; Pugacheva, E.; Klenova, E.; Grundy, P.; et al. Tumor-associated zinc finger mutations in the CTCF transcription factor selectively alter tts DNA-binding specificity. Cancer Res. 2002, 62, 48–52. [Google Scholar] [PubMed]
- Wolfe, S.A.; Nekludova, L.; Pabo, C.O. DNA recognition by Cys2His2 zinc finger proteins. Ann. Rev. Biophys. Biomol. Struct. 2000, 29, 183–212. [Google Scholar] [CrossRef]
- Marshall, A.D.; Bailey, C.G.; Rasko, J.E. CTCF and BORIS in genome regulation and cancer. Curr. Opin. Genet. Dev. 2014, 24, 8–15. [Google Scholar] [CrossRef]
- Braccioli, L.; de Wit, E. CTCF: A Swiss-army knife for genome organization and transcription regulation. Essays Biochem. 2019, 63, 157–165. [Google Scholar] [CrossRef]
- Davidson, I.F.; Bauer, B.; Goetz, D.; Tang, W.; Wutz, G.; Peters, J.M. DNA loop extrusion by human cohesin. Science 2019, 366, 1338–1345. [Google Scholar] [CrossRef]
- Liu, F.; Wu, D.; Wang, X. Roles of CTCF in conformation and functions of chromosome. Semin. Cell Dev. Biol. 2019, 90, 168–173. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, P.; Wang, L. Many facades of CTCF unified by its coding for three-dimensional genome architecture. J. Genet. Genom. 2020, 47, 407–424. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]
- Kemp, C.J.; Moore, J.M.; Moser, R.; Bernard, B.; Teater, M.; Smith, L.E.; Rabaia, N.A.; Gurley, K.E.; Guinney, J.; Busch, S.E.; et al. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 2014, 7, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Marshall, A.D.; Bailey, C.G.; Champ, K.; Vellozzi, M.; O’Young, P.; Metierre, C.; Feng, Y.; Thoeng, A.; Richards, A.M.; Schmitz, U.; et al. CTCF genetic alterations in endometrial carcinoma are pro-tumorigenic. Oncogene 2017, 36, 4100–4110. [Google Scholar] [CrossRef] [Green Version]
- Debaugny, R.E.; Skok, J.A. CTCF and CTCFL in cancer. Curr. Opin. Genet. Dev. 2020, 61, 44–52. [Google Scholar] [CrossRef]
- Yin, M.; Wang, J.; Wang, M.; Li, X.; Zhang, M.; Wu, Q.; Wang, Y. Molecular mechanism of directional CTCF recognition of a diverse range of genomic sites. Cell Res. 2017, 27, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.J.; Miranda, M.A.; O’Hern, M.J.; McElroy, J.P.; Coombes, K.R.; Bundschuh, R.; Cohn, D.E.; Mutch, D.G.; Goodfellow, P.J. Patterns of CTCF and ZFHX3 Mutation and Associated Outcomes in Endometrial Cancer. J. Natl. Cancer Inst. 2015, 107, djv249. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; Park, M.j.; Terui, K.; Suzuki, H.; et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 2013, 45, 1293–1299. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Molecular Lesions of Insulator CTCF and Its Paralogue CTCFL (BORIS) in Cancer: An Analysis from Published Genomic Studies. High-Throughput 2018, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Le Gallo, M.; O’Hara, A.J.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; O’Neil, N.J.; Price, J.C.; Zhang, S.; England, B.M.; Godwin, A.K.; et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 2012, 44, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Schlick, T. Molecular Modeling and Simulation: An Interdisciplinary Guide, 2nd ed.; Antman, S.S., Marsden, J.E., Sirovich, L., Eds.; Springer: New York, NY, USA, 2010. [Google Scholar]
- Bailey, C.G.; Gupta, S.; Metierre, C.; Amarasekera, P.M.S.; O’Young, P.; Kyaw, W.; Laletin, T.; Francis, H.; Semaan, C.; Sharifi Tabar, M.; et al. Structure-function relationships explain CTCF zinc finger mutation phenotypes in cancer. Cell. Mol. Life Sci. 2021, 78, 7519–7536. [Google Scholar] [CrossRef]
- Konrad, E.D.H.; Nardini, N.; Caliebe, A.; Nagel, I.; Young, D.; Horvath, G.; Santoro, S.L.; Shuss, C.; Ziegler, A.; Bonneau, D.; et al. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet. Med. 2019, 21, 2723–2733. [Google Scholar] [CrossRef] [Green Version]
- Holwerda, S.J.B.; de Laat, W. CTCF: The protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Tian, K.; Huang, J.; Duan, W.; Fu, H.; Feng, Y.; Wang, H.; Jiang, Y.; Li, Y.; Wang, R.; et al. CTCF DNA-binding domain undergoes dynamic and selective protein–protein interactions. iScience 2022, 25, 105011. [Google Scholar] [CrossRef] [PubMed]
- Katainen, R.; Dave, K.; Pitkänen, E.; Palin, K.; Kivioja, T.; Välimäki, N.; Gylfe, A.E.; Ristolainen, H.; Hänninen, U.A.; Cajuso, T.; et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 2015, 47, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Monahan, K.; Wu, H.; Gertz, J.; Varley, K.E.; Li, W.; Myers, R.M.; Maniatis, T.; Wu, Q. CTCF/cohesin-mediated DNA looping is required for protocadherin promoter choice. Proc. Natl. Acad. Sci. USA 2012, 109, 21081–21086. [Google Scholar] [CrossRef] [Green Version]
- Portillo-Ledesma, S.; Li, Z.; Schlick, T. Genome modeling: From chromatin fibers to genes. Curr. Opin. Struct. Biol. 2023, 78, 102506. [Google Scholar] [CrossRef]
- Chan, K.L.; Bakman, I.; Marts, A.R.; Batir, Y.; Dowd, T.L.; Tierney, D.L.; Gibney, B.R. Characterization of the Zn(II) Binding Properties of the Human Wilms’ Tumor Suppressor Protein C-terminal Zinc Finger Peptide. Inorg. Chem. 2014, 53, 6309–6320. [Google Scholar] [CrossRef] [Green Version]
- Sénèque, O.; Latour, J.M. Coordination Properties of Zinc Finger Peptides Revisited: Ligand Competition Studies Reveal Higher Affinities for Zinc and Cobalt. J. Am. Chem. Soc. 2010, 132, 17760–17774. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Merz , K.M.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Pang, Y.P.; Xu, K.; Yazal, J.E.; Prendergast, F.G. Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 2000, 9, 1857–1865. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building water models: A different approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E.I.I.I. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC.: New York, NY, USA, 2015.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | R339Q | R342C | S354T | K365T | R377H | R377C | Q418R | R448Q |
|---|---|---|---|---|---|---|---|---|
| −73 ± 11 | −53 ± 5 | −81 ± 10 | −72 ± 13 | −82 ± 16 | −65 ± 12 | −69 ± 10 | −69 ± 7 | −61 ± 15 |
| ZF | WT | R339Q | R342C | S354T | K365T | R377H | R377C | Q418R | R448Q |
|---|---|---|---|---|---|---|---|---|---|
| (ZF3) | (ZF3) | (ZF4) | (ZF4) | (ZF4–5) | (ZF4–5) | (ZF6) | (ZF7) | ||
| 3 | 3 | 0 | 4 | 1 | 3 | 2 | 3 | 4 | 0 |
| 4 | 6 | 2 | 4 | 1 | 4 | 0 | 0 | 3 | 0 |
| 5 | 4 | 4 | 3 | 2 | 3 | 3 | 3 | 4 | 3 |
| 6 | 4 | 6 | 8 | 6 | 5 | 6 | 5 | 7 | 5 |
| 7 | 1 | 2 | 4 | 2 | 2 | 1 | 4 | 0 | 0 |
| Total | 18 | 14 | 23 | 12 | 17 | 12 | 15 | 18 | 8 |
| Bond | k (kcal/mol Å2) | Req (Å) | |
|---|---|---|---|
| DZ-ZN | 540 | 0.90 | |
| DZ-DZ | 540 | 1.47 | |
| Angle | k (kcal/mol radian2) | Teq (deg.) | |
| DZ-ZN-DZ | 55 | 109.50 | |
| DZ-DZ-DZ | 55 | 60.0 | |
| DZ-DZ-ZN | 55 | 35.25 | |
| Dihedral | IDIVF | Vn/2 (kcal/mol) | γ (deg.) N |
| ZN-DZ-DZ-DZ | 1 | 0 | 35.3 |
| DZ-ZN-DZ-DZ | 1 | 0 | 120.0 |
| DZ-DZ-DZ-DZ | 1 | 0 | 70.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, A.; Chen, C.; Portillo-Ledesma, S.; Schlick, T. Effect of Single-Residue Mutations on CTCF Binding to DNA: Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2023, 24, 6395. https://doi.org/10.3390/ijms24076395
Mao A, Chen C, Portillo-Ledesma S, Schlick T. Effect of Single-Residue Mutations on CTCF Binding to DNA: Insights from Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2023; 24(7):6395. https://doi.org/10.3390/ijms24076395
Chicago/Turabian StyleMao, Albert, Carrie Chen, Stephanie Portillo-Ledesma, and Tamar Schlick. 2023. "Effect of Single-Residue Mutations on CTCF Binding to DNA: Insights from Molecular Dynamics Simulations" International Journal of Molecular Sciences 24, no. 7: 6395. https://doi.org/10.3390/ijms24076395