
Involvement of Bcl-2 Family Proteins in Tetraploidization-Related Senescence
 , ,
, ,
Abstract
:1. Introduction
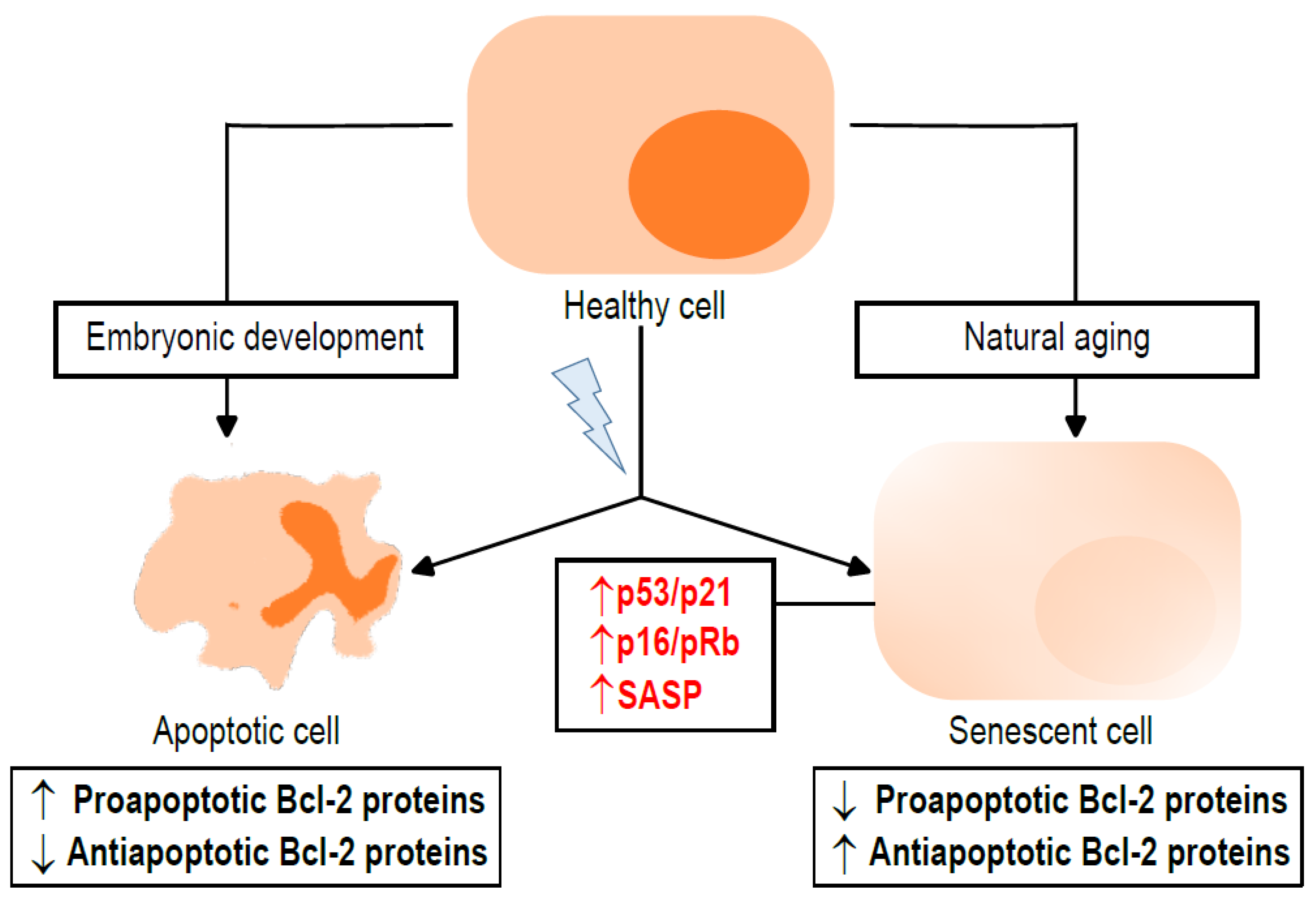
2. Apoptosis and Senescence: Two Ways to Suppress Cell Proliferation
3. Role of the Bcl-2 Family in Senescence
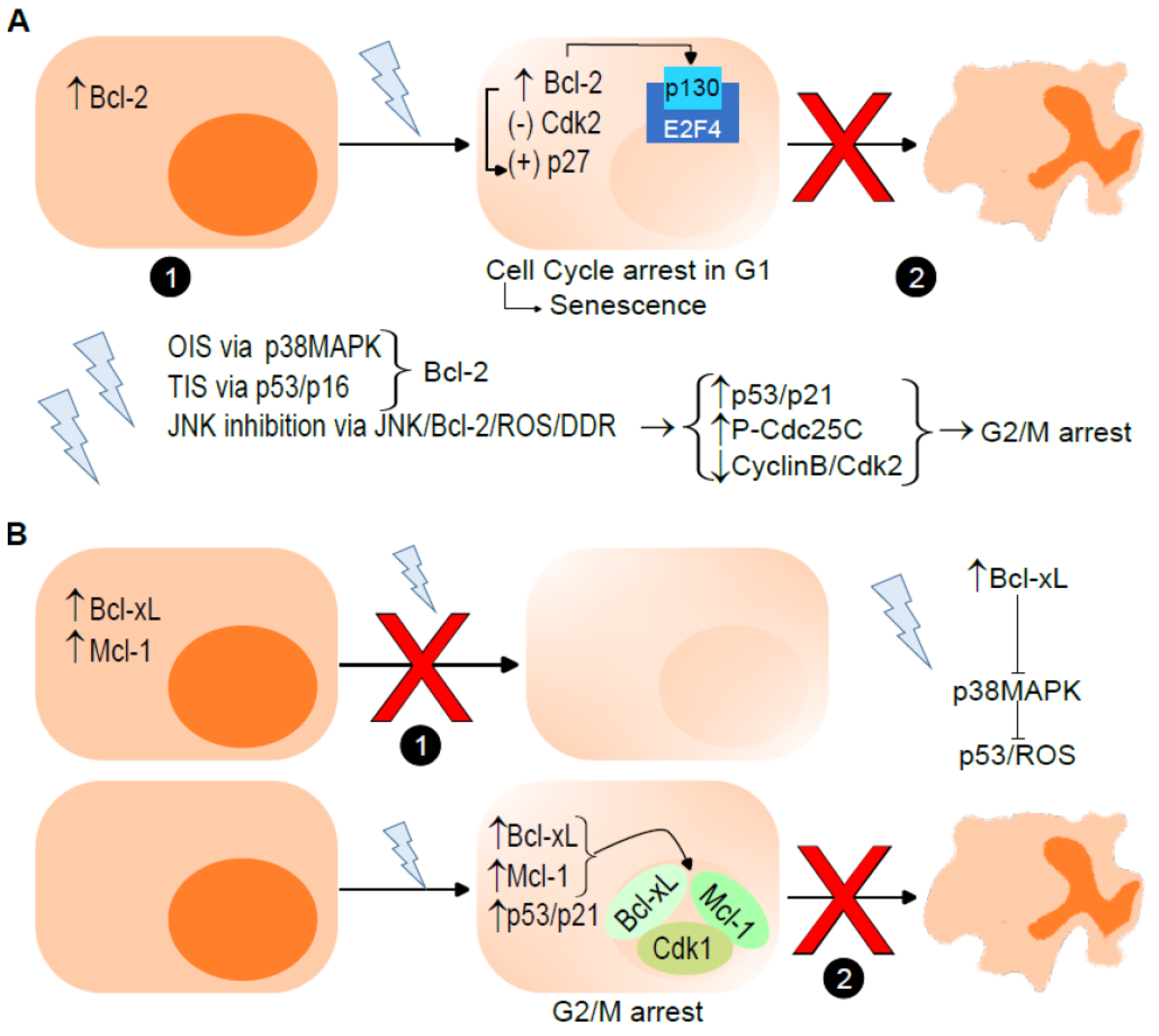
3.1. Antiapoptotic Bcl-2 Proteins
3.2. Multidomain Proapoptotic Bcl-2 Proteins
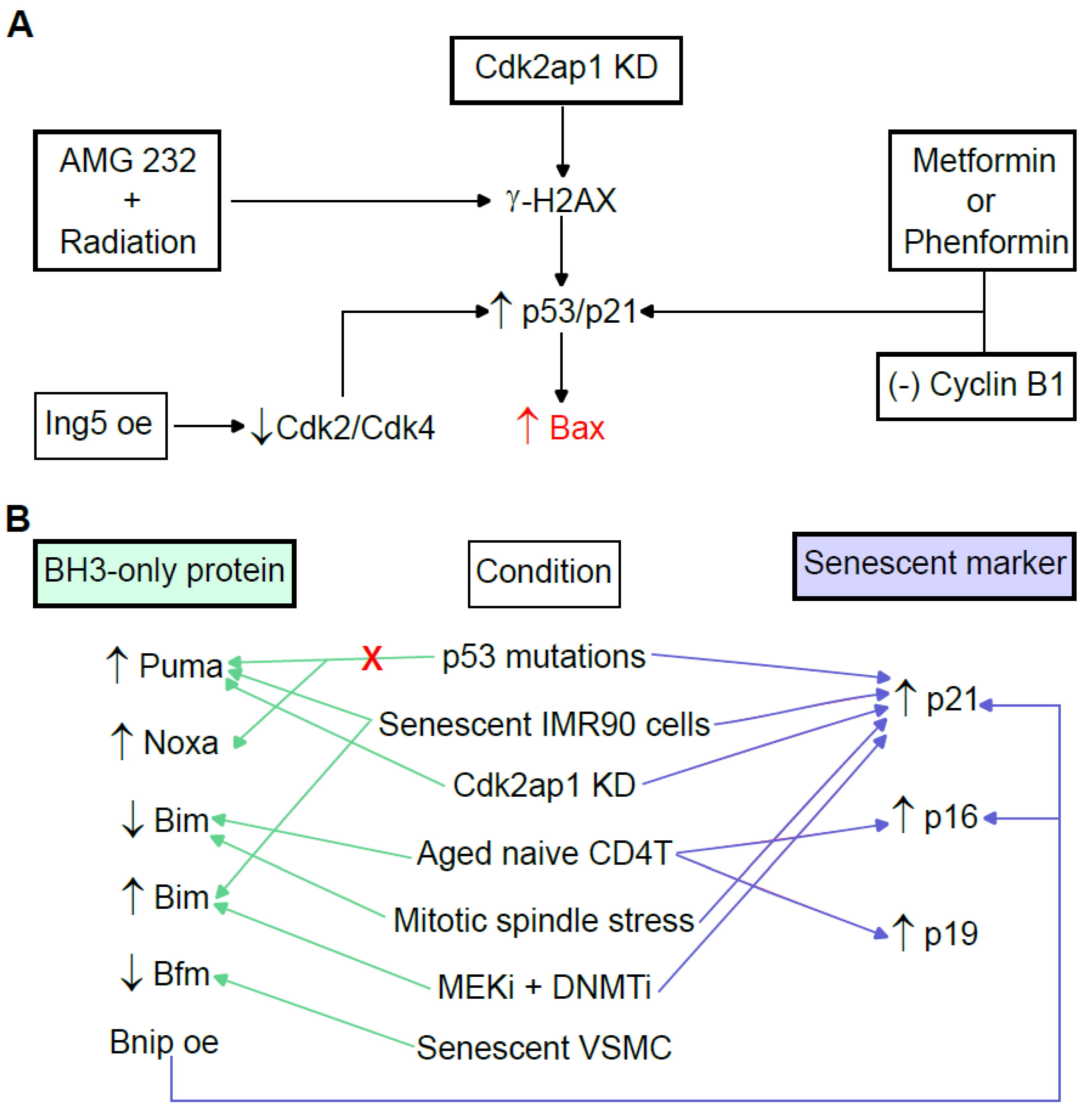
3.3. BH3-Only Proapoptotic Bcl-2 Proteins
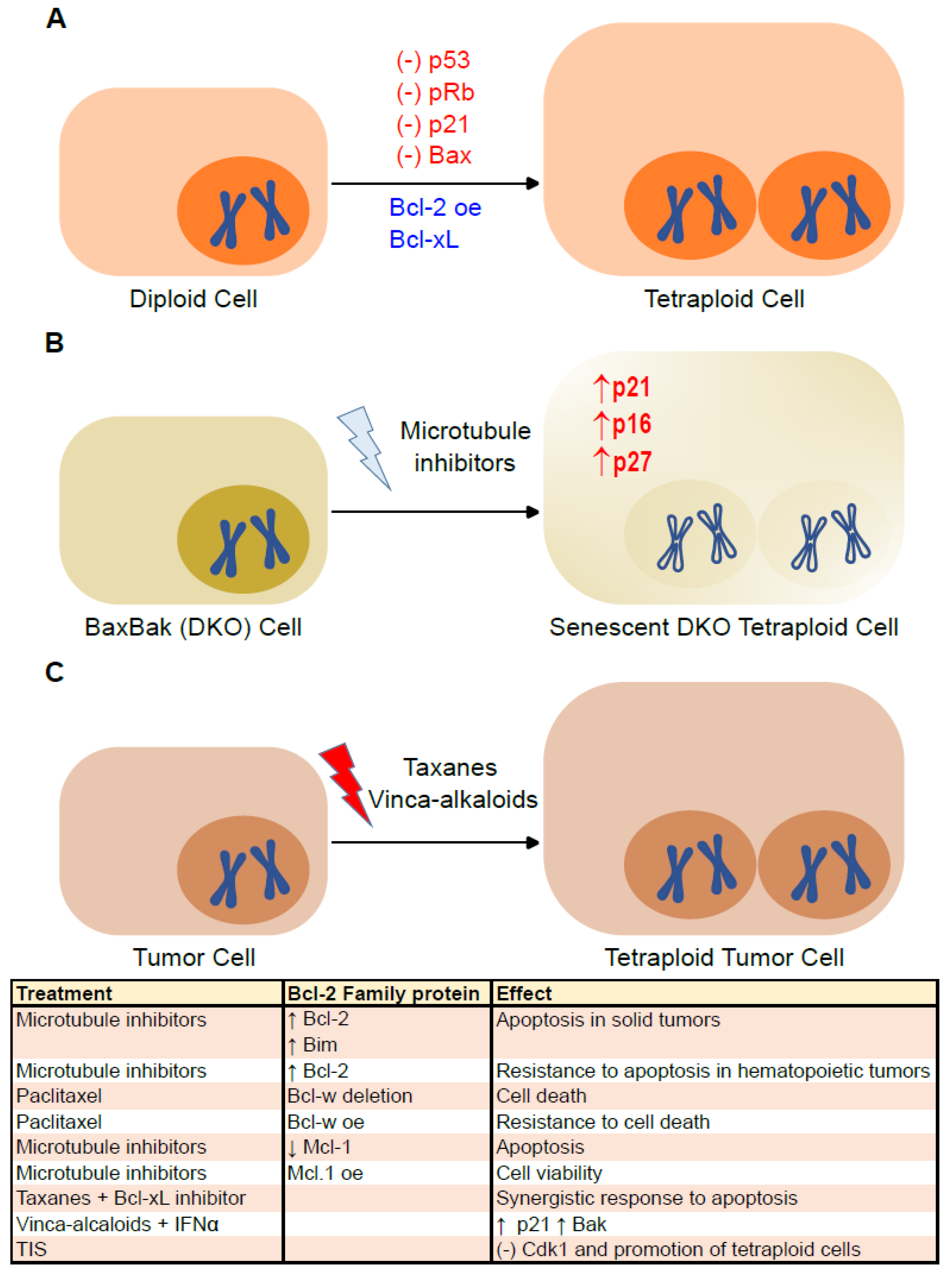
4. Senescence and Bcl-2 Family Proteins in Tetraploid Cells
5. The Bcl-2 Family Proteins as a Target for Senolytic Agents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy + Senolytic Agent + Other Agents | Disease | Target/s | Mode of Action | References |
|---|---|---|---|---|
| ABT-199 | Bcl-2 | Releases Bax from Bcl-2 inhibition | ||
| ABT-263 | Bcl-2 | Inhibition of Bcl-2-Bcl-xL interaction | ||
| Bcl-w | Release of Bim | |||
| Bcl-xL | Bax translocation | |||
| Apoptosis | ||||
| Irradiation + ABT-199 or ABT-263 | Soft tissue sarcomas | Therapy-Induced Senescence | [93] | |
| Apoptosis | ||||
| Gemcitabine + Everolimus or Ionizing radiation + ABT-263 | Malignant meningioma | Senescence | [94,95] | |
| Apoptosis | ||||
| Wogonin + ABT-263 | T-cell malignancies | p53-mediated DNA damage | [96] | |
| Upregulation of Bcl-2 | ||||
| Apoptosis | ||||
| ABT-199 + Bortezomib | Soft tissue sarcomas | Release of Bax from Bcl-2 inhibition | [97] | |
| Accumulation of Bok and Noxa | ||||
| Inhibition of Mcl-1 | ||||
| Apoptosis | ||||
| ABT-263 | Senescent HUVECs | Senolytic effect | [98] | |
| IMR90 cells | ||||
| Senescent MuSCs | ||||
| DNA damage inducers + ABT-263 | Prostate cancer-TIS | Interferes with the interaction between Bcl-xL and Bax | [100] | |
| Apoptosis | ||||
| ABT-263 | Breast cancer Tp53+/+ | [101] | ||
| ABT-737 | Bcl-2 | Inhibits the protective effect of Bcl-2 and Bcl-xL | [23,28,102] | |
| Bcl-w | activate the cleavage of caspases 8/9 | |||
| Bcl-xL | Mechanism of action has not been described in detail | |||
| ABT-737 | PanIn lesions | Eliminates Cox2-expressing senescent cells | [103] | |
| TMZ + ABT-737 or ABT-263 | Senescent glioblastoma cells | Senolytic effect | [104] | |
| Radiation+ TMZ + Bcl-xL inhibitors (ABT-263, A1331852, A1155463) | GBM | Increases the vulnerability of GBM to TMZ treatment | [43] | |
| TMZ + ABT-199 | GBM | Senolytic effect | [105] | |
| Dox + A1331852 | Xenograft models of solid tumors | Disruption of Bcl-xL:Bim complexes | [108,109] | |
| Induces cytochrome c release | ||||
| Activation caspases 3/7 | ||||
| Externalization of phosphatidylserine | ||||
| Apoptosis | ||||
| A1331852 | Bid | Upregulate the expression of Bid and Bax | [22] | |
| Bax | ||||
| Ionizing radiation + A1331852 or A1155463 | HUVECs | Induces cleavage of caspase 3/7 | [110] | |
| IMR90 cells | ||||
| A1210477 | Mcl-1 | Mcl-1 inhibitors | [111] | |
| S63845 | ||||
| A1210477 + EE-84 | Chronic myelogenous leukemia | Synergistic effect | [111] | |
| S63845 | Senescent tumor cells and metastases | Complete elimination | [50] | |
| A1210477 | Myeloma | Disrupt Mcl-1/Bak complexes | [112] | |
| Apoptosis | ||||
| Taxanes + Vinca alkaloids + Combination of antiapoptotic proteins of the BCL-2 family inhibitors | Increases the efficacy of microtubule inhibitors | [52] | ||
| Nintedanib | Triple-negative breast cancer cells | Tyrosine kinase inhibitor | Induces apoptosis | [115,116,117,119] |
| Malignant pleural mesothelioma | Inhibits tumor growth | |||
| Non-small cell lung cancer | Bim expression | |||
| Cleavage of Caspase-9 and caspases 3/7 | ||||
| P5091 | USP7 inhibitor | Promotes ubiquitination and degradation of Mdm2 | [120] | |
| Increases p53, Puma and Noxa | ||||
| Inhibits the interaction of Bcl-xL and Bak | ||||
| Apoptosis in senescent cells |
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chong, S.J.F.; Marchi, S.; Petroni, G.; Kroemer, G.; Galluzzi, L.; Pervaiz, S. Noncanonical Cell Fate Regulation by Bcl-2 Proteins. Trends Cell Biol. 2020, 30, 537–555. [Google Scholar] [CrossRef]
- Basu, A. The interplay between apoptosis and cellular senescence: Bcl-2 family proteins as targets for cancer therapy. Pharmacol. Ther. 2022, 230, 107943. [Google Scholar] [CrossRef]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent Telomere Damage Induces Bypass of Mitosis and Tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Dumontet, C.; Sikic, B.I. Mechanisms of Action of and Resistance to Antitubulin Agents: Microtubule Dynamics, Drug Transport, and Cell Death. J. Clin. Oncol. 1999, 17, 1061. [Google Scholar] [CrossRef] [Green Version]
- Manchado, E.; Guillamot, M.; Malumbres, M. Killing cells by targeting mitosis. Cell Death Differ. 2012, 19, 369–377. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005, 8, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Rieder, C.L.; Maiato, H. Stuck in division or passing through: What happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Valent, A.; Penault-Llorca, F.; Cayre, A.; Kroemer, G. Change in HER2 (ERBB2) gene status after taxane-based chemotherapy for breast cancer: Polyploidization can lead to diagnostic pitfalls with potential impact for clinical management. Cancer Genet. 2013, 206, 37–41. [Google Scholar] [CrossRef]
- Malaquin, N.; Vancayseele, A.; Gilbert, S.; Antenor-Habazac, L.; Olivier, M.A.; Ait Ali Brahem, Z.; Saad, F.; Delouya, G.; Rodier, F. DNA Damage- But Not Enzalutamide-Induced Senescence in Prostate Cancer Promotes Senolytic Bcl-xL Inhibitor Sensitivity. Cells 2020, 9, 1593. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef]
- Barille-Nion, S.; Bah, N.; Véquaud, E.; Juin, P. Regulation of cancer cell survival by BCL2 family members upon prolonged mitotic arrest: Opportunities for anticancer therapy. Anticancer. Res. 2012, 32, 4225–4233. [Google Scholar] [PubMed]
- Deng, J.; Gutiérrez, L.G.; Stoll, G.; Motiño, O.; Martins, I.; Núñez, L.; Pedro, J.M.B.-S.; Humeau, J.; Bordenave, C.; Pan, J.; et al. Paradoxical implication of BAX/BAK in the persistence of tetraploid cells. Cell Death Dis. 2021, 12, 1039. [Google Scholar] [CrossRef] [PubMed]
- Rysanek, D.; Vasicova, P.; Kolla, J.N.; Sedlak, D.; Andera, L.; Bartek, J.; Hodny, Z. Synergism of BCL-2 family inhibitors facilitates selective elimination of senescent cells. Aging 2022, 14, 6381–6414. [Google Scholar] [CrossRef]
- Domingos, P.M.; Steller, H. Pathways regulating apoptosis during patterning and development. Curr. Opin. Genet. Dev. 2007, 17, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [Green Version]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Yang, Y.; Mihajlovic, M.; Valentijn, F.; Nguyen, T.Q.; Goldschmeding, R.; Masereeuw, R. A Human Conditionally Immortalized Proximal Tubule Epithelial Cell Line as a Novel Model for Studying Senescence and Response to Senolytics. Front. Pharmacol. 2022, 13, 791612. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Jagadish, N.; Surolia, A.; Suri, A. Heat shock protein 70-2 (HSP70-2) a novel cancer testis antigen that promotes growth of ovarian cancer. Am. J. Cancer Res. 2017, 7, 1252–1269. [Google Scholar] [PubMed]
- Wu, G.; Li, X.; Zhan, Y.; Fan, X.; Xu, L.; Chen, T.; Wang, X. BID- and BAX-mediated mitochondrial pathway dominates A-1331852-induced apoptosis in senescent A549 cells. Biochem. Biophys. Res. Commun. 2022, 627, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Litwiniec, A.; Gackowska, L.; Helmin-Basa, A.; Żuryń, A.; Grzanka, A. Low-dose etoposide-treatment induces endoreplication and cell death accompanied by cytoskeletal alterations in A549 cells: Does the response involve senescence? The possible role of vimentin. Cancer Cell Int. 2013, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Breger, K.S.; Smith, L.; Turker, M.S.; Thayer, M.J. Ionizing Radiation Induces Frequent Translocations with Delayed Replication and Condensation. Cancer Res. 2004, 64, 8231–8238. [Google Scholar] [CrossRef] [Green Version]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; Van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Cheng, J.; Zeng, H.; Shao, L. Senescent Cell Depletion Through Targeting BCL-Family Proteins and Mitochondria. Front. Physiol. 2020, 11, 593630. [Google Scholar] [CrossRef]
- Drullion, C.; Trégoat, C.; Lagarde, V.; Tan, S.; Gioia, R.; Priault, M.; Djavaheri-Mergny, M.; Brisson, A.A.; Auberger, P.; Mahon, F.-X.; et al. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis. 2012, 3, e373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincheval, V.; Renaud, F.; Lemaire, C.; Godefroy, N.; Trotot, P.; Boulo, V.; Mignotte, B.; Vayssiere, J.L. Bcl-2 can promote p53-dependent senescence versus apoptosis without affecting the G1/S transition. Biochem. Biophys. Res. Commun. 2002, 298, 282–288. [Google Scholar] [CrossRef] [PubMed]
- López-Diazguerrero, N.E.; López-Araiza, H.; Conde-Perezprina, J.C.; Bucio, L.; Cárdenas-Aguayo, M.C.; Ventura, J.L.; Covarrubias, L.; Gutiérrez-Ruíz, M.C.; Zentella, A.; Königsberg, M. Bcl-2 protects against oxidative stress while inducing premature senescence. Free. Radic. Biol. Med. 2006, 40, 1161–1169. [Google Scholar] [CrossRef]
- Sasaki, M.; Kumazaki, T.; Takano, H.; Nishiyama, M.; Mitsui, Y. Senescent cells are resistant to death despite low Bcl-2 level. Mech. Ageing Dev. 2001, 122, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Vairo, G.; Soos, T.J.; Upton, T.M.; Zalvide, J.; DeCaprio, J.A.; Ewen, M.E.; Koff, A.; Adams, J.M. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol. Cell. Biol. 2000, 20, 4745–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelyudova, A.; Aksenov, N.D.; Pospelov, V.; Pospelova, T. By Blocking Apoptosis, Bcl-2 in p38-Dependent Manner Promotes Cell Cycle Arrest and Accelerated Senescence After DNA Damage and Serum Withdrawal. Cell Cycle 2007, 6, 2171–2177. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, D.; Li, H. Initiation of premature senescence by Bcl-2 in hypoxic condition. Int. J. Clin. Exp. Pathol. 2014, 7, 2446–2453. [Google Scholar]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A Senescence Program Controlled by p53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Lee, J.H.; Ko, Y.G.; Hong, S.I.; Lee, J.S. Prevention of premature senescence requires JNK regulation of Bcl-2 and reactive oxygen species. Oncogene 2010, 29, 561–575. [Google Scholar] [CrossRef] [Green Version]
- Jemaà, M.; Vitale, I.; Kepp, O.; Berardinelli, F.; Galluzzi, L.; Senovilla, L.; Mariño, G.; Malik, S.A.; Rello-Varona, S.; Lissa, D.; et al. Selective killing of p53-deficient cancer cells by SP600125. EMBO Mol. Med. 2012, 4, 500–514. [Google Scholar] [CrossRef]
- Kim, J.A.; Lee, J.; Margolis, R.L.; Fotedar, R. SP600125 suppresses Cdk1 and induces endoreplication directly from G2 phase, independent of JNK inhibition. Oncogene 2010, 29, 1702–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.Y.; Shin, H.J.; Bae, I.H. miR-93-5p suppresses cellular senescence by directly targeting Bcl-w and p21. Biochem. Biophys. Res. Commun. 2018, 505, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Borrás, C.; Mas-Bargues, C.; Román-Domínguez, A.; Sanz-Ros, J.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From C. elegans to Human Centenarians. Int. J. Mol. Sci. 2020, 21, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selt, F.; Sigaud, R.; Valinciute, G.; Sievers, P.; Zaman, J.; Alcon, C.; Schmid, S.; Peterziel, H.; Tsai, J.W.; Guiho, R.; et al. BH3 mimetics targeting BCL-XL impact the senescent compartment of pilocytic astrocytoma. Neuro-Oncol. 2022, noac199. [Google Scholar] [CrossRef]
- Ikezawa, K.; Hikita, H.; Shigekawa, M.; Iwahashi, K.; Eguchi, H.; Sakamori, R.; Tatsumi, T.; Takehara, T. Increased Bcl-xL Expression in Pancreatic Neoplasia Promotes Carcinogenesis by Inhibiting Senescence and Apoptosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 185–200.e1. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.S.; Jin, D.H.; Chae, H.D.; Kang, S.; Kim, S.C.; Bang, Y.J.; Choi, T.S.; Choi, K.S.; Shin, D.Y. Bcl-xL and E1B-19K proteins inhibit p53-induced irreversible growth arrest and senescence by preventing reactive oxygen species-dependent p38 activation. J. Biol. Chem. 2004, 279, 17765–17771. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, E.; Beauchemin, M.; Bertrand, R. Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) during G2/M cell-cycle checkpoint. Oncogene 2007, 26, 5851–5865. [Google Scholar] [CrossRef] [Green Version]
- Hayward, R.L.; MacPherson, J.S.; Cummings, J.; Monia, B.P.; Smyth, J.F.; Jodrell, D.I. Antisense Bcl-xl down-regulation switches the response to topoisomerase I inhibition from senescence to apoptosis in colorectal cancer cells, enhancing global cytotoxicity. Clin. Cancer Res. 2003, 9, 2856–2865. [Google Scholar]
- Tsuji, K.; Kida, Y.; Koshikawa, N.; Yamamoto, S.; Shinozaki, Y.; Watanabe, T.; Lin, J.; Nagase, H.; Takenaga, K. Suppression of non-small-cell lung cancer A549 tumor growth by an mtDNA mutation-targeting pyrrole-imidazole polyamide-triphenylphosphonium and a senolytic drug. Cancer Sci. 2022, 113, 1321–1337. [Google Scholar] [CrossRef]
- Rahman, M.; Olson, I.; Mansour, M.; Carlstrom, L.P.; Sutiwisesak, R.; Saber, R.; Rajani, K.; Warrington, A.E.; Howard, A.; Schroeder, M.; et al. Selective Vulnerability of Senescent Glioblastoma Cells to BCL-XL Inhibition. Mol. Cancer Res. 2022, 20, 938–948. [Google Scholar] [CrossRef]
- de Carne Trecesson, S.; Guillemin, Y.; Belanger, A.; Bernard, A.C.; Preisser, L.; Ravon, E.; Gamelin, E.; Juin, P.; Barre, B.; Coqueret, O. Escape from p21-mediated oncogene-induced senescence leads to cell dedifferentiation and dependence on anti-apoptotic Bcl-xL and MCL1 proteins. J. Biol. Chem. 2011, 286, 12825–12838. [Google Scholar] [CrossRef] [Green Version]
- Bolesta, E.; Pfannenstiel, L.W.; Demelash, A.; Lesniewski, M.L.; Tobin, M.; Schlanger, S.E.; Nallar, S.C.; Papadimitriou, J.C.; Kalvakolanu, D.V.; Gastman, B.R. Inhibition of Mcl-1 Promotes Senescence in Cancer Cells: Implications for Preventing Tumor Growth and Chemotherapy Resistance. Mol. Cell. Biol. 2012, 32, 1879–1892. [Google Scholar] [CrossRef] [Green Version]
- Troiani, M.; Colucci, M.; D’Ambrosio, M.; Guccini, I.; Pasquini, E.; Varesi, A.; Valdata, A.; Mosole, S.; Revandkar, A.; Attanasio, G.; et al. Single-cell transcriptomics identifies Mcl-1 as a target for senolytic therapy in cancer. Nat. Commun. 2022, 13, 2177. [Google Scholar] [CrossRef]
- Jamil, S.; Sobouti, R.; Hojabrpour, P.; Raj, M.; Kast, J.; Duronio, V. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem. J. 2005, 387, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Whitaker, R.H.; Placzek, W.J. Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents. Cells 2019, 8, 346. [Google Scholar] [CrossRef] [Green Version]
- Demelash, A.; Pfannenstiel, L.W.; Tannenbaum, C.S.; Li, X.; Kalady, M.F.; DeVecchio, J.; Gastman, B.R. Structure-Function Analysis of the Mcl-1 Protein Identifies a Novel Senescence-regulating Domain. J. Biol. Chem. 2015, 290, 21962–21975. [Google Scholar] [CrossRef] [Green Version]
- Demelash, A.; Pfannenstiel, L.W.; Liu, L.; Gastman, B.R. Mcl-1 regulates reactive oxygen species via NOX4 during chemotherapy-induced senescence. Oncotarget 2017, 8, 28154–28168. [Google Scholar] [CrossRef] [Green Version]
- Forman, K.; Vara, E.; García, C.; Kireev, R.; Cuesta, S.; Acuña-Castroviejo, D.; Tresguerres, J.A.F. Beneficial effects of melatonin on cardiological alterations in a murine model of accelerated aging. J. Pineal Res. 2010, 49, 312–320. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone Modifications in Senescence-Associated Resistance to Apoptosis by Oxidative Stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Alsayegh, K.N.; Gadepalli, V.S.; Iyer, S.; Rao, R.R. Knockdown of CDK2AP1 in Primary Human Fibroblasts Induces p53 Dependent Senescence. PLoS ONE 2015, 10, e0120782. [Google Scholar] [CrossRef] [Green Version]
- Werner, L.R.; Huang, S.; Francis, D.M.; Armstrong, E.A.; Ma, F.; Li, C.; Iyer, G.; Canon, J.; Harari, P.M. Small Molecule Inhibition of MDM2–p53 Interaction Augments Radiation Response in Human Tumors. Mol. Cancer Ther. 2015, 14, 1994–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Zhao, M.; Parris, A.B.; Feng, X.; Yang, X. p53 is required for metformin-induced growth inhibition, senescence and apoptosis in breast cancer cells. Biochem. Biophys. Res. Commun. 2015, 464, 1267–1274. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Li, X.; Meng, W.; Bai, Z.; Rui, S.; Wang, Z.; Zhou, W.; Jin, X. Effect of CCNB1 silencing on cell cycle, senescence, and apoptosis through the p53 signaling pathway in pancreatic cancer. J. Cell. Physiol. 2018, 234, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Zhao, Z.-J.; He, H.-Y.; Wu, J.-C.; Ding, X.-Q.; Yang, L.; Jia, N.; Li, Z.-J.; Zheng, H.-C. The roles of ING5 in gliomas: A good marker for tumorigenesis and a potential target for gene therapy. Oncotarget 2017, 8, 56558–56568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haschka, M.; Karbon, G.; Fava, L.L.; Villunger, A. Perturbing mitosis for anti-cancer therapy: Is cell death the only answer? EMBO Rep. 2018, 19, e45440. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Huston, G.E.; Dibble, J.; Duso, D.K.; Swain, S.L. Bim Dictates Naive CD4 T Cell Lifespan and the Development of Age-Associated Functional Defects. J. Immunol. 2010, 185, 4535–4544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Schneider, L.; Essmann, F.; Cirstea, I.C.; Kuck, F.; Kletke, A.; Jänicke, R.U.; Wiek, C.; Hanenberg, H.; Ahmadian, M.R.; et al. The centrosomal protein TACC3 controls paclitaxel sensitivity by modulating a premature senescence program. Oncogene 2010, 29, 6184–6192. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, J.; Emmons, M.F.; Faião-Flores, F.; Aplin, A.E.; Harbour, J.W.; Licht, J.D.; Wink, M.R.; Smalley, K.S.M.; Avezedo, J.G. Decitabine limits escape from MEK inhibition in uveal melanoma. Pigment. Cell Melanoma Res. 2020, 33, 507–514. [Google Scholar] [CrossRef]
- Zhang, J.; Patel, J.M.; Block, E.R. Enhanced apoptosis in prolonged cultures of senescent porcine pulmonary artery endothelial cells. Mech. Ageing Dev. 2002, 123, 613–625. [Google Scholar] [CrossRef]
- Stickles, X.B.; Marchion, D.C.; Bicaku, E.; AL Sawah, E.; Abbasi, F.; Xiong, Y.; Zgheib, N.B.; Boac, B.M.; Orr, B.C.; Judson, P.L.; et al. BAD-mediated apoptotic pathway is associated with human cancer development. Int. J. Mol. Med. 2015, 35, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Zhan, J.K.; Zhong, J.Y.; Wang, Y.J.; Wang, Y.; Li, S.; He, J.Y.; Tan, P.; Chen, Y.Y.; Liu, X.B.; et al. lncRNA-ES3/miR-34c-5p/BMF axis is involved in regulating high-glucose-induced calcification/senescence of VSMCs. Aging 2019, 11, 523–535. [Google Scholar] [CrossRef]
- Zhuang, L.; Ly, R.; Rösl, F.; Niebler, M. p53 Is Regulated in a Biphasic Manner in Hypoxic Human Papillomavirus Type 16 (HPV16)-Positive Cervical Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9533. [Google Scholar] [CrossRef]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis, via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.I.; Jo, E.-R.; Song, H. Urolithin A attenuates auditory cell senescence by activating mitophagy. Sci. Rep. 2022, 12, 7704. [Google Scholar] [CrossRef]
- Boveri, T. The Origin of Malignant Tumors; Williams & Wilkins: Baltimore, MD, USA, 1929. [Google Scholar]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef]
- Was, H.; Borkowska, A.; Olszewska, A.; Klemba, A.; Marciniak, M.; Synowiec, A.; Kieda, C. Polyploidy formation in cancer cells: How a Trojan horse is born. Semin. Cancer Biol. 2022, 81, 24–36. [Google Scholar] [CrossRef]
- Margolis, R.L.; Lohez, O.D.; Andreassen, P.R. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell. Biochem. 2003, 88, 673–683. [Google Scholar] [CrossRef]
- Panopoulos, A.; Pacios-Bras, C.; Choi, J.; Yenjerla, M.; Sussman, M.A.; Fotedar, R.; Margolis, R.L. Failure of cell cleavage induces senescence in tetraploid primary cells. Mol. Biol. Cell 2014, 25, 3105–3118. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Bloukh, S.; Gewirtz, D.A. Targeting tumor cell senescence and polyploidy as potential therapeutic strategies. Semin. Cancer Biol. 2022, 81, 37–47. [Google Scholar] [CrossRef]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [Green Version]
- Castedo, M.; Coquelle, A.; Vivet, S.; Vitale, I.; Kauffmann, A.; Dessen, P.; Pequignot, M.O.; Casares, N.; Valent, A.; Mouhamad, S.; et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006, 25, 2584–2595. [Google Scholar] [CrossRef] [Green Version]
- Senovilla, L.; Vitale, I.; Martins, I.; Tailler, M.; Pailleret, C.; Michaud, M.; Galluzzi, L.; Adjemian, S.; Kepp, O.; Niso-Santano, M.; et al. An Immunosurveillance Mechanism Controls Cancer Cell Ploidy. Science 2012, 337, 1678–1684. [Google Scholar] [CrossRef]
- Boilève, A.; Senovilla, L.; Vitale, I.; Lissa, D.; Martins, I.; Métivier, D.; van den Brink, S.; Clevers, H.; Galluzzi, L.; Castedo, M.; et al. Immunosurveillance against tetraploidization-induced colon tumorigenesis. Cell Cycle 2013, 12, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Aranda, F.; Chaba, K.; Bloy, N.; Garcia, P.; Bordenave, C.; Martins, I.; Stoll, G.; Tesniere, A.; Kroemer, G.; Senovilla, L. Immune effectors responsible for the elimination of hyperploid cancer cells. Oncoimmunology 2018, 7, e1463947. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Tang, Y.; Wang, R.; Najafi, M. Mechanisms of cancer cell death induction by paclitaxel: An updated review. Apoptosis 2022, 27, 647–667. [Google Scholar] [CrossRef]
- Ogden, A.; Rida, P.C.; Knudsen, B.S.; Kucuk, O.; Aneja, R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett. 2015, 367, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Upreti, M.; Koonce, N.A.; Hennings, L.; Chambers, T.C.; Griffin, R.J. Pegylated IFN-α sensitizes melanoma cells to chemotherapy and causes premature senescence in endothelial cells by IRF-1-mediated signaling. Cell Death Dis. 2010, 1, e67. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat. Rev. Drug Discov. 2018, 17, 377. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-C.; Kim, J.-R. Senotherapeutics: Emerging strategy for healthy aging and age-related disease. BMB Rep. 2019, 52, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2022, 290, 1362–1383. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Lafontaine, J.; Cardin, G.; Malaquin, N.; Boisvert, J.-S.; Rodier, F.; Wong, P. Senolytic Targeting of Bcl-2 Anti-Apoptotic Family Increases Cell Death in Irradiated Sarcoma Cells. Cancers 2021, 13, 386. [Google Scholar] [CrossRef]
- Yamamoto, M.; Suzuki, S.; Togashi, K.; Sugai, A.; Okada, M.; Kitanaka, C. Gemcitabine Cooperates with Everolimus to Inhibit the Growth of and Sensitize Malignant Meningioma Cells to Apoptosis Induced by Navitoclax, an Inhibitor of Anti-Apoptotic BCL-2 Family Proteins. Cancers 2022, 14, 1706. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sanomachi, T.; Suzuki, S.; Togashi, K.; Sugai, A.; Seino, S.; Sato, A.; Okada, M.; Kitanaka, C. Gemcitabine radiosensitization primes irradiated malignant meningioma cells for senolytic elimination by navitoclax. Neuro-Oncol. Adv. 2021, 3, vdab148. [Google Scholar] [CrossRef]
- Qing, Y.; Li, H.; Zhao, Y.; Hu, P.; Wang, X.; Yu, X.; Zhu, M.; Wang, H.; Wang, Z.; Guo, Q.; et al. One-Two Punch Therapy for the Treatment of T-Cell Malignancies Involving p53-Dependent Cellular Senescence. Oxidative Med. Cell. Longev. 2021, 2021, 5529518. [Google Scholar] [CrossRef]
- Muenchow, A.; Weller, S.; Hinterleitner, C.; Malenke, E.; Bugl, S.; Wirths, S.; Müller, M.R.; Schulze-Osthoff, K.; Aulitzky, W.E.; Kopp, H.-G.; et al. The BCL-2 selective inhibitor ABT-199 sensitizes soft tissue sarcomas to proteasome inhibition by a concerted mechanism requiring BAX and NOXA. Cell Death Dis. 2020, 11, 701. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.D.; Souers, A.J.; Alotaibi, M.R.; Faber, A.C.; Reed, J.; Harada, H.; et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X(L) -BAX interaction. Mol. Oncol. 2020, 14, 2504–2519. [Google Scholar] [CrossRef] [PubMed]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ. 2020, 27, 3097–3116. [Google Scholar] [CrossRef]
- Kline, M.P.; Rajkumar, S.V.; Timm, M.M.; Kimlinger, T.K.; Haug, J.L.; Lust, J.A.; Greipp, P.R.; Kumar, S. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia 2007, 21, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin-Gal, D.; Roitman, L.; Ovadya, Y.; Azazmeh, N.; Assouline, B.; Schlesinger, Y.; Kalifa, R.; Horwitz, S.; Khalatnik, Y.; Hochner-Ger, A.; et al. Senolytic elimination of Cox2-expressing senescent cells inhibits the growth of premalignant pancreatic lesions. Gut 2022, 71, 345–355. [Google Scholar] [CrossRef]
- Beltzig, L.; Christmann, M.; Kaina, B. Abrogation of Cellular Senescence Induced by Temozolomide in Glioblastoma Cells: Search for Senolytics. Cells 2022, 11, 2588. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, C.; Tatsch, L.; Vilar, J.B.; Rasenberger, B.; Beltzig, L.; Kaina, B.; Tomicic, M.; Christmann, M. Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers 2021, 13, 3585. [Google Scholar] [CrossRef] [PubMed]
- Moncsek, A.; Al-Suraih, M.S.; Trussoni, C.E.; O’Hara, S.P.; Splinter, P.L.; Zuber, C.; Patsenker, E.; Valli, P.V.; Fingas, C.D.; Weber, A.; et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2(−/−)) mice. Hepatology 2018, 67, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Zhang, C.; Xu, L.; Chen, H.; Fan, X.; Sun, B.; Tang, Q.; Zhan, Y.; Chen, T.; Wang, X. BAK plays a key role in A-1331852-induced apoptosis in senescent chondrocytes. Biochem. Biophys. Res. Commun. 2022, 609, 93–99. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef]
- Wang, L.; Doherty, G.A.; Judd, A.S.; Tao, Z.F.; Hansen, T.M.; Frey, R.R.; Song, X.; Bruncko, M.; Kunzer, A.R.; Wang, X.; et al. Discovery of A-1331852, a First-in-Class, Potent, and Orally-Bioavailable BCL-X(L) Inhibitor. ACS Med. Chem. Lett. 2020, 11, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Kim, S.; El-Sawy, E.; Cerella, C.; Orlikova-Boyer, B.; Kirsch, G.; Christov, C.; Dicato, M.; Diederich, M. Anti-Leukemic Properties of Aplysinopsin Derivative EE-84 Alone and Combined to BH3 Mimetic A-1210477. Mar. Drugs 2021, 19, 285. [Google Scholar] [CrossRef] [PubMed]
- Bougie, P.G.; Maiga, S.; Tessoulin, B.; Bourcier, J.; Bonnet, A.; Rodriguez, M.S.; Le Gouill, S.; Touzeau, C.; Moreau, P.; Pellat-Deceunynck, C.; et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood 2018, 132, 2656–2669. [Google Scholar] [CrossRef] [Green Version]
- Senichkin, V.V.; Pervushin, N.V.; Zamaraev, A.V.; Sazonova, E.V.; Zuev, A.P.; Streletskaia, A.Y.; Prikazchikova, T.A.; Zatsepin, T.S.; Kovaleva, O.V.; Tchevkina, E.M.; et al. Bak and Bcl-xL Participate in Regulating Sensitivity of Solid Tumor Derived Cell Lines to Mcl-1 Inhibitors. Cancers 2021, 14, 181. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Huang, T.-T.; Chu, P.-Y.; Huang, C.-T.; Lee, C.-H.; Wang, W.-L.; Lau, K.-Y.; Tsai, W.-C.; Chao, T.-I.; Su, J.-C.; et al. The tyrosine kinase inhibitor nintedanib activates SHP-1 and induces apoptosis in triple-negative breast cancer cells. Exp. Mol. Med. 2017, 49, e366. [Google Scholar] [CrossRef] [Green Version]
- Laszlo, V.; Valko, Z.; Kovacs, I.; Ozsvar, J.; Hoda, M.A.; Klikovits, T.; Lakatos, D.; Czirok, A.; Garay, T.; Stiglbauer, A.; et al. Nintedanib Is Active in Malignant Pleural Mesothelioma Cell Models and Inhibits Angiogenesis and Tumor Growth In Vivo. Clin. Cancer Res. 2018, 24, 3729–3740. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, T.; Tanaka, H.; Tabe, C.; Tsuchiya, J.; Ishioka, Y.; Itoga, M.; Taima, K.; Takanashi, S.; Tasaka, S. Effect of nintedanib on non-small cell lung cancer in a patient with idiopathic pulmonary fibrosis: A case report and literature review. Thorac. Cancer 2020, 11, 1720–1723. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Shin, Y.-J.; Palumbo, S.; Papageorgiou, I.; Hahn, S.; Irish, J.D.; Rounseville, S.P.; Krafty, R.T.; Wollin, L.; Sauler, M.; et al. Leveraging ageing models of pulmonary fibrosis: The efficacy of nintedanib in ageing. Eur. Respir. J. 2021, 58, 2100759. [Google Scholar] [CrossRef]
- Cho, H.-J.; Hwang, J.-A.; Yang, E.J.; Kim, E.-C.; Kim, J.-R.; Kim, S.Y.; Kim, Y.Z.; Park, S.C.; Lee, Y.-S. Nintedanib induces senolytic effect via STAT3 inhibition. Cell Death Dis. 2022, 13, 760. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, W.; Lv, D.; Zhang, X.; Zhang, X.; Ortiz, Y.T.; Budamagunta, V.; Campisi, J.; Zheng, G.; Zhou, D. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell 2020, 19, e13117. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barriuso, D.; Alvarez-Frutos, L.; Gonzalez-Gutierrez, L.; Motiño, O.; Kroemer, G.; Palacios-Ramirez, R.; Senovilla, L. Involvement of Bcl-2 Family Proteins in Tetraploidization-Related Senescence. Int. J. Mol. Sci. 2023, 24, 6374. https://doi.org/10.3390/ijms24076374
Barriuso D, Alvarez-Frutos L, Gonzalez-Gutierrez L, Motiño O, Kroemer G, Palacios-Ramirez R, Senovilla L. Involvement of Bcl-2 Family Proteins in Tetraploidization-Related Senescence. International Journal of Molecular Sciences. 2023; 24(7):6374. https://doi.org/10.3390/ijms24076374
Chicago/Turabian StyleBarriuso, Daniel, Lucia Alvarez-Frutos, Lucia Gonzalez-Gutierrez, Omar Motiño, Guido Kroemer, Roberto Palacios-Ramirez, and Laura Senovilla. 2023. "Involvement of Bcl-2 Family Proteins in Tetraploidization-Related Senescence" International Journal of Molecular Sciences 24, no. 7: 6374. https://doi.org/10.3390/ijms24076374