
Mechanisms of Demyelination and Remyelination Strategies for Multiple Sclerosis

Abstract
:1. Introduction
2. Roles of Peripheral and CNS Resident Innate Immune Cells
3. Roles of Oligodendrocyte Lineage Cells in Remyelination
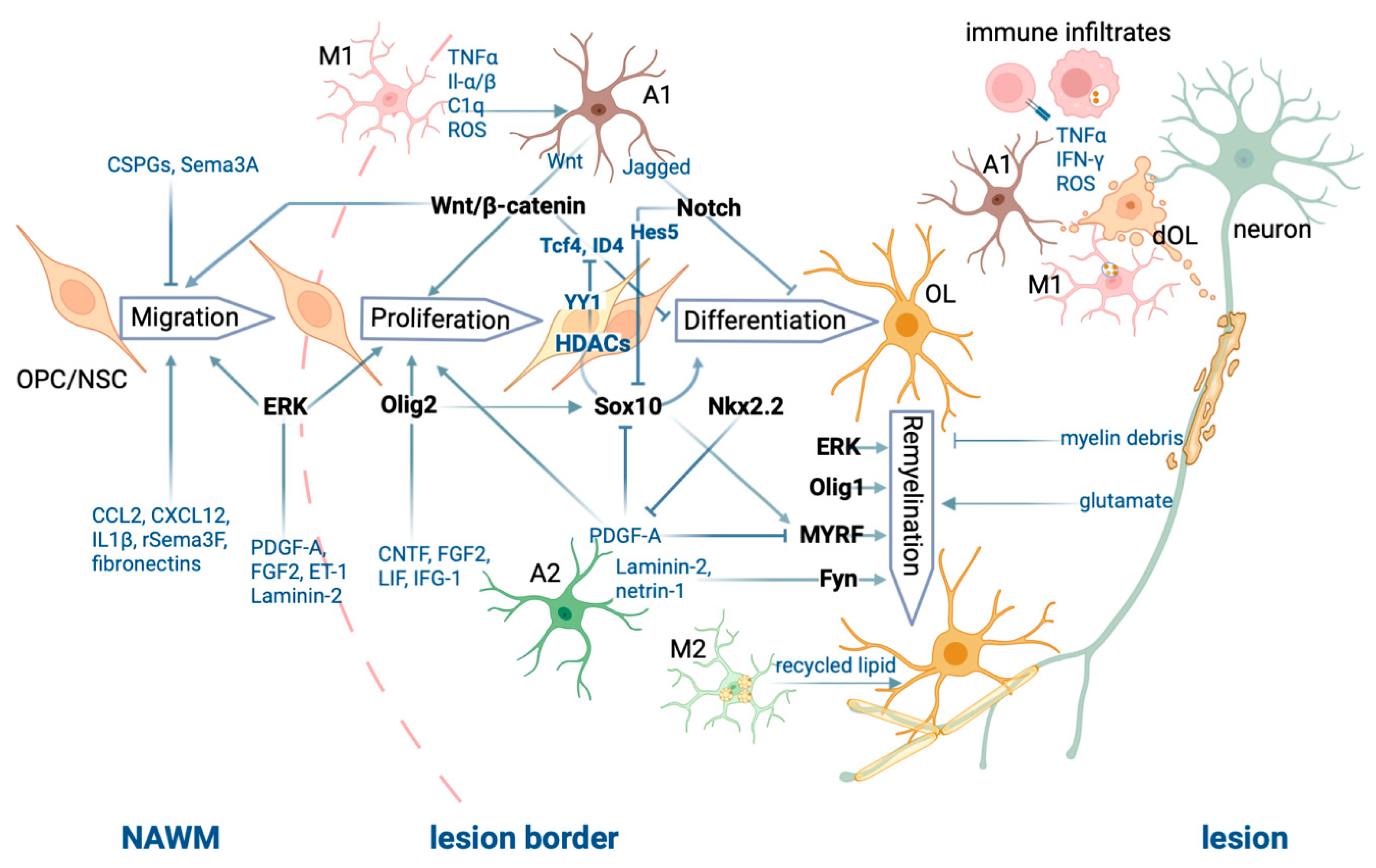
3.1. Oligodendrocyte Progenitor Cells Migrate to Lesion Sites and Proliferate Following CNS Injury
3.2. Key Transcription Factors for Oligodendrocyte Maturation and (Re)myelination
3.3. Epigenetic Modulation of Myelination in Oligodendrocyte Lineage Cells and the Aging Process
4. Treatment Perspectives for Remyelination
4.1. Agents That Directly Promote OPC Differentiation and Remyelination
4.2. Rejuvenating Aged OPCs to Repopulate Remyelinating OLs
4.3. Preventing OPC Differentiation Arrest and OL Death Mediated by an Inflamed Environment
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hövelmeyer, N.; Hao, Z.; Kranidioti, K.; Kassiotis, G.; Buch, T.; Frommer, F.; von Hoch, L.; Kramer, D.; Minichiello, L.; Kollias, G.; et al. Apoptosis of oligodendrocytes via FAS and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J. Immunol. 2005, 175, 5875–5884. [Google Scholar] [CrossRef] [Green Version]
- Waisman, A.; Hauptmann, J.; Regen, T. The role of IL-17 in CNS diseases. Acta Neuropathol. 2015, 129, 625–637. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Krämer-Albers, E.-M. Extracellular vesicles in the oligodendrocyte microenvironment. Neurosci. Lett. 2020, 725, 134915. [Google Scholar] [CrossRef]
- Reynolds, R.; Roncaroli, F.; Nicholas, R.; Radotra, B.; Gveric, D.; Howell, O. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol. 2011, 122, 155–170. [Google Scholar] [CrossRef]
- Duncan, I.D.; Marik, R.L.; Broman, A.T.; Heidari, M. Thin myelin sheaths as the hallmark of remyelination persist over time and preserve axon function. Proc. Natl. Acad. Sci. USA 2017, 114, E9685–E9691. [Google Scholar] [CrossRef] [Green Version]
- Stangel, M.; Kuhlmann, T.; Matthews, P.M.; Kilpatrick, T.J. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 742–754. [Google Scholar] [CrossRef]
- Bodini, B.; Veronese, M.; García-Lorenzo, D.; Battaglini, M.; Poirion, E.; Chardain, A.; Freeman, L.; Louapre, C.; Tchikviladze, M.; Papeix, C.; et al. Dynamic Imaging of Individual Remyelination Profiles in Multiple Sclerosis. Ann. Neurol. 2016, 79, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.F.E.; Cunniffe, N.G.; Franklin, R.J.M. Seeing is believing: Identifying remyelination in the Central Nervous System. Curr. Opin. Pharmacol. 2022, 66, 102269. [Google Scholar] [CrossRef] [PubMed]
- Heß, K.; Starost, L.; Kieran, N.W.; Thomas, C.; Vincenten, M.C.; Antel, J.; Martino, G.; Huitinga, I.; Healy, L.; Kuhlmann, T. Lesion stage-dependent causes for impaired remyelination in MS. Acta Neuropathol. 2020, 140, 359–375. [Google Scholar] [CrossRef]
- Van den Bosch, A.M.; Hümmert, S.; Steyer, A.; Ruhwedel, T.; Hamann, J.; Smolders, J.; Nave, K.A.; Stadelmann, C.; Kole, M.H.; Möbius, W.; et al. Ultrastructural axon–myelin unit alterations in multiple sclerosis correlate with inflammation. Ann. Neurol. 2023. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.G.; Wegner, C.; Kreutzfeldt, M.; Neid, K.; Thal, D.R.; Jürgens, T.; Brück, W.; Stadelmann, C.; Merkler, D. Oligodendroglia in cortical multiple sclerosis lesions decrease with disease progression, but regenerate after repeated experimental demyelination. Acta Neuropathol. 2014, 128, 231–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B.; et al. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 403–422. [Google Scholar] [CrossRef] [PubMed]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin fat facts: An overview of lipids and fatty acid metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giussani, P.; Prinetti, A.; Tringali, C. The role of sphingolipids in myelination and myelin stability and their involvement in childhood and adult demyelinating disorders. J. Neurochem. 2020, 156, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.G.; Zheng, B. Myelin-associated inhibitors in axonal growth after CNS injury. Curr. Opin. Neurobiol. 2014, 27, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Winzeler, A.M.; Mandemakers, W.J.; Sun, M.Z.; Stafford, M.; Phillips, C.T.; Barres, B.A. The lipid sulfatide is a novel myelin-associated inhibitor of CNS axon outgrowth. J. Neurosci. 2011, 31, 6481–6492. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.J.M.; Simons, M. CNS remyelination and inflammation: From basic mechanisms to therapeutic opportunities. Neuron 2022, 110, 3549–3565. [Google Scholar] [CrossRef] [PubMed]
- De Rosbo, N.K.; Hoffman, M.; Mendel, I.; Yust, I.; Kaye, J.; Bakimer, R.; Flechter, S.; Abramsky, O.; Milo, R.; Karni, A.; et al. Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: Reactivity to the extracellular domain of Mog is directed against three main regions. Eur. J. Immunol. 1997, 27, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Ortega, S.B.; Noorbhai, I.; Poinsatte, K.; Kong, X.; Anderson, A.; Monson, N.L.; Stowe, A.M. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov. Med. 2015, 106, 381–392. [Google Scholar]
- Fadok, V.A.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.; Henson, P.M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000, 405, 85–90. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagórska, A.; Través, P.G.; Schlessinger, J.; Lemke, G. Author response: Differential Tam receptor–ligand–phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3, e03385. [Google Scholar] [CrossRef]
- Shen, K.; Reichelt, M.; Kyauk, R.V.; Ngu, H.; Shen, Y.-A.A.; Foreman, O.; Modrusan, Z.; Friedman, B.A.; Sheng, M.; Yuen, T.J. Multiple sclerosis risk gene Mertk is required for microglial activation and subsequent remyelination. Cell Rep. 2021, 34, 108835. [Google Scholar] [CrossRef]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef]
- Gouna, G.; Klose, C.; Bosch-Queralt, M.; Liu, L.; Gokce, O.; Schifferer, M.; Cantuti-Castelvetri, L.; Simons, M. TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J. Exp. Med. 2021, 218, e20210227. [Google Scholar] [CrossRef]
- Xie, M.; Liu, Y.; Zhao, S.; Zhang, L.; Bosco, D.; Pang, Y.-P.; Sheth, U.; Martens, Y.; Zhao, N.; Liu, C.-C.; et al. TREM2 interacts with TDP-43 and mediates microglial neuroprotection against TDP-43-related neurodegeneration. Nat. Neurosci. 2021, 25, 26–38. [Google Scholar] [CrossRef]
- Deerhake, M.E.; Biswas, D.D.; Barclay, W.E.; Shinohara, M.L. Pattern recognition receptors in multiple sclerosis and its animal models. Front. Immunol. 2019, 10, 2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J. Clin. Investig. 2006, 116, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Hernandez, S.; Gerlach, N.; Fletcher, J.M.; Biros, E.; Mack, M.; Körner, H.; Baxter, A.G. Role for Myd88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 791–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, M.I.; Su, M.; Cantuti-Castelvetri, L.; Müller, S.A.; Schifferer, M.; Djannatian, M.; Alexopoulos, I.; van der Meer, F.; Winkler, A.; van Ham, T.J.; et al. Pro-inflammatory activation following demyelination is required for myelin clearance and oligodendrogenesis. J. Exp. Med. 2020, 217, e20191390. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Düking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2020, 24, 47–60. [Google Scholar] [CrossRef]
- Mailleux, J.; Vanmierlo, T.; Bogie, J.F.J.; Wouters, E.; Lütjohann, D.; Hendriks, J.J.A.; van Horssen, J. Active liver X receptor signaling in phagocytes in multiple sclerosis lesions. Mult. Scler. 2017, 24, 279–289. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.-T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective Cholesterol clearance limits remyelination in the aged Central Nervous System. Science 2018, 359, 684–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Toyama, Y.; Nishiyama, A. Differentiation of proliferated ng2-positive glial progenitor cells in a remyelinating lesion. J. Neurosci. Res. 2002, 69, 826–836. [Google Scholar] [CrossRef]
- Nishiyama, A.; Lin, X.-H.; Giese, N.; Heldin, C.-H.; Stallcup, W.B. Co-localization of NG2 proteoglycan and PDGF-alpha-receptor on O2A progenitor cells in the developing rat brain. J. Neurosci. Res. 1996, 43, 299–314. [Google Scholar] [CrossRef]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the Adult Brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef] [Green Version]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Höglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-Van Evercooren, A. Activation of the subventricular zone in multiple sclerosis: Evidence for early glial progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.J.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.L.; Roth, P.T.; Stratton, J.A.; Chuang, B.H.; Danne, J.; Ellis, S.L.; Ng, S.W.; Kilpatrick, T.J.; Merson, T.D. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. 2014, 34, 14128–14146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, J.; Grund, E.M.; Silva, H.M.; Lafaille, J.J.; Fishell, G.; Salzer, J.L. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 2015, 526, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyon, S.; Dubessy, A.L.; Aigrot, M.S.; Trotter, M.; Huang, J.K.; Dauphinot, L.; Potier, M.C.; Kerninon, C.; Melik Parsadaniantz, S.; Franklin, R.J.; et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J. Neurosci. 2015, 35, 4–20. [Google Scholar] [CrossRef] [Green Version]
- Antel, J.P.; Lin, Y.H.; Cui, Q.-L.; Pernin, F.; Kennedy, T.E.; Ludwin, S.K.; Healy, L.M. Immunology of oligodendrocyte precursor cells in vivo and in vitro. J. Neuroimmunol. 2019, 331, 28–35. [Google Scholar] [CrossRef]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and Mouse Models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Sharma, K.; Frost, E.E.; Pillai, P.P. Role of PDGF-A-activated ERK signaling mediated FAK-paxillin interaction in oligodendrocyte progenitor cell migration. J. Mol. Neurosci. 2019, 67, 564–573. [Google Scholar] [CrossRef]
- Hoshina, N.; Tezuka, T.; Yokoyama, K.; Kozuka-Hata, H.; Oyama, M.; Yamamoto, T. Focal adhesion kinase regulates laminin-induced oligodendroglial process outgrowth. Genes Cells 2007, 12, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- De Jong, J.M.; Wang, P.; Oomkens, M.; Baron, W. Remodeling of the interstitial extracellular matrix in white matter multiple sclerosis lesions: Implications for remyelination (failure). J. Neurosci. Res. 2020, 98, 1370–1397. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Tran, A.P.; Xin, L.; Sanapala, C.; Lang, B.T.; Silver, J.; Yang, Y. Modulation of proteoglycan receptor ptpσ enhances MMP-2 activity to promote recovery from multiple sclerosis. Nat. Commun. 2018, 9, 4126. [Google Scholar] [CrossRef] [Green Version]
- Foerster, S.; Neumann, B.; McClain, C.; Di Canio, L.; Chen, C.Z.; Reich, D.S.; Simons, B.D.; Franklin, R.J.M. Proliferation is a requirement for differentiation of oligodendrocyte progenitor cells during CNS remyelination. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through Notch Activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadea, A.; Aguirre, A.; Haydar, T.F.; Gallo, V. Endothelin-1 regulates oligodendrocyte development. J. Neurosci. 2009, 29, 10047–10062. [Google Scholar] [CrossRef] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [Green Version]
- Fancy, S.P.J.; Baranzini, S.E.; Zhao, C.; Yuk, D.-I.; Irvine, K.-A.; Kaing, S.; Sanai, N.; Franklin, R.J.M.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Kagawa, T.; Wada, T.; Muroyama, Y.; Takada, S.; Ikenaka, K. Wnt signaling controls the timing of oligodendrocyte development in the spinal cord. Dev. Biol. 2005, 282, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.-H.; Niu, J.; Munji, R.; Davalos, D.; Chang, J.; Zhang, H.; Tien, A.-C.; Kuo, C.J.; Chan, J.R.; Daneman, R.; et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 2016, 351, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in Vertebrate Neural Development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef]
- Lee, H.K.; Laug, D.; Zhu, W.; Patel, J.M.; Ung, K.; Arenkiel, B.R.; Fancy, S.P.; Mohila, C.; Deneen, B. APCDD1 stimulates oligodendrocyte differentiation after white matter injury. Glia 2015, 63, 1840–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genoud, S.; Lappe-Siefke, C.; Goebbels, S.; Radtke, F.; Aguet, M.; Scherer, S.S.; Suter, U.; Nave, K.-A.; Mantei, N. Notch1 control of oligodendrocyte differentiation in the spinal cord. J. Cell Biol. 2002, 158, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Argaw, A.T.; Gurfein, B.T.; Zameer, A.; Snyder, B.J.; Ge, C.; Lu, Q.R.; Rowitch, D.H.; Raine, C.S.; Brosnan, C.F.; et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc. Natl. Acad. Sci. USA 2009, 106, 19162–19167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stidworthy, M.F. Notch1 and Jagged1 are expressed after CNS demyelination, but are not a major rate-determining factor during remyelination. Brain 2004, 127, 1928–1941. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Li, J.; Marin-Husstege, M.; Kageyama, R.; Fan, Y.; Gelinas, C.; Casaccia-Bonnefil, P. A molecular insight of hes5-dependent inhibition of myelin gene expression: Old partners and New Players. EMBO J. 2006, 25, 4833–4842. [Google Scholar] [CrossRef]
- Fancy, S.P.J.; Zhao, C.; Franklin, R.J.M. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol. Cell. Neurosci. 2004, 27, 247–254. [Google Scholar] [CrossRef]
- Ligon, K.L.; Kesari, S.; Kitada, M.; Sun, T.; Arnett, H.A.; Alberta, J.A.; Anderson, D.J.; Stiles, C.D.; Rowitch, D.H. Development of NG2 neural progenitor cells requires Olig gene function. Proc. Natl. Acad. Sci. USA 2006, 103, 7853–7858. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zuo, H.; Maher, B.J.; Serwanski, D.R.; LoTurco, J.J.; Lu, Q.R.; Nishiyama, A. Olig2-dependent developmental fate switch of Ng2 Cells. Development 2012, 139, 2299–2307. [Google Scholar] [CrossRef] [Green Version]
- Wegener, A.; Deboux, C.; Bachelin, C.; Frah, M.; Kerninon, C.; Seilhean, D.; Weider, M.; Wegner, M.; Nait-Oumesmar, B. Gain of OLIG2 function in oligodendrocyte progenitors promotes remyelination. Brain 2014, 138, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Arnett, H.A.; Fancy, S.P.; Alberta, J.A.; Zhao, C.; Plant, S.R.; Kaing, S.; Raine, C.S.; Rowitch, D.H.; Franklin, R.J.; Stiles, C.D. BHLH transcription factor OLIG1 is required to repair demyelinated lesions in the CNS. Science 2004, 306, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhao, X.; Zheng, K.; Li, H.; Huang, H.; Zhang, Z.; Mastracci, T.; Wegner, M.; Chen, Y.; Sussel, L.; et al. Genetic evidence that Nkx2.2 and Pdgfra are major determinants of the timing of oligodendrocyte differentiation in the developing CNS. Development 2014, 141, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sock, E.; Wegner, M. Using the lineage determinants Olig2 and Sox10 to explore transcriptional regulation of oligodendrocyte development. Dev. Neurobiol. 2021, 81, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Hornig, J.; Fröb, F.; Vogl, M.R.; Hermans-Borgmeyer, I.; Tamm, E.R.; Wegner, M. The transcription factors SOX10 and MYRF define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet. 2013, 9, e1003907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, M.; Vaquié, A.; Nocera, G.; Heller, M.; Stumpe, M.; Siva Sankar, D.; Dengjel, J.; Meijer, D.; Yamaguchi, T.; Matthias, P.; et al. EEF1A1 deacetylation enables transcriptional activation of remyelination. Nat. Commun. 2020, 11, 3420. [Google Scholar] [CrossRef]
- Fyffe-Maricich, S.L.; Karlo, J.C.; Landreth, G.E.; Miller, R.H. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J. Neurosci. 2011, 31, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Ishii, A.; Fyffe-Maricich, S.L.; Furusho, M.; Miller, R.H.; Bansal, R. Erk1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J. Neurosci. 2012, 32, 8855–8864. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Ferner, A.H.; Wong, A.W.; Denham, M.; Kilpatrick, T.J.; Murray, S.S. Extracellular signal-regulated kinase ½ signaling promotes oligodendrocyte myelination in vitro. J. Neurochem. 2012, 122, 1167–1180. [Google Scholar] [CrossRef]
- Fyffe-Maricich, S.L.; Schott, A.; Karl, M.; Krasno, J.; Miller, R.H. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J. Neurosci. 2013, 33, 18402–18408. [Google Scholar] [CrossRef] [Green Version]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Jeffries, M.A.; Urbanek, K.; Torres, L.; Wendell, S.G.; Rubio, M.E.; Fyffe-Maricich, S.L. ERK1/2 activation in preexisting oligodendrocytes of adult mice drives new myelin synthesis and enhanced CNS function. J. Neurosci. 2016, 36, 9186–9200. [Google Scholar] [CrossRef] [PubMed]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef] [Green Version]
- Emery, B.; Agalliu, D.; Cahoy, J.D.; Watkins, T.A.; Dugas, J.C.; Mulinyawe, S.B.; Ibrahim, A.; Ligon, K.L.; Rowitch, D.H.; Barres, B.A. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 2009, 138, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Koenning, M.; Jackson, S.; Hay, C.M.; Faux, C.; Kilpatrick, T.J.; Willingham, M.; Emery, B. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J. Neurosci. 2012, 32, 12528–12542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aprato, J.; Sock, E.; Weider, M.; Elsesser, O.; Fröb, F.; Wegner, M. MYRF guides target gene selection of transcription factor SOX10 during oligodendroglial development. Nucleic Acids Res. 2019, 48, 1254–1270. [Google Scholar] [CrossRef] [PubMed]
- Krämer-Albers, E.-M.; White, R. From axon–glial signalling to myelination: The integrating role of oligodendroglial Fyn Kinase. Cell. Mol. Life Sci. 2011, 68, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Gonsior, C.; Krämer-Albers, E.-M.; Stöhr, N.; Hüttelmaier, S.; Trotter, J. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2–dependent RNA granules. J. Cell Biol. 2008, 181, 579–586. [Google Scholar] [CrossRef]
- Jepson, S.; Vought, B.; Gross, C.H.; Gan, L.; Austen, D.; Frantz, J.D.; Zwahlen, J.; Lowe, D.; Markland, W.; Krauss, R. Lingo-1, a transmembrane signaling protein, inhibits oligodendrocyte differentiation and myelination through intercellular self-interactions. J. Biol. Chem. 2012, 287, 22184–22195. [Google Scholar] [CrossRef] [Green Version]
- Rajasekharan, S.; Baker, K.A.; Horn, K.E.; Jarjour, A.A.; Antel, J.P.; Kennedy, T.E. Netrin 1 and DCC regulate oligodendrocyte process branching and membrane extension via Fyn and Rhoa. Development 2009, 136, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Relucio, J.; Tzvetanova, I.D.; Ao, W.; Lindquist, S.; Colognato, H. Laminin alters Fyn regulatory mechanisms and promotes oligodendrocyte development. J. Neurosci. 2009, 29, 11794–11806. [Google Scholar] [CrossRef] [Green Version]
- Samudyata; Castelo-Branco, G.; Liu, J. Epigenetic regulation of oligodendrocyte differentiation: From development to demyelinating disorders. Glia 2020, 68, 1619–1630. [Google Scholar] [CrossRef]
- Koreman, E.; Sun, X.; Lu, Q.R. Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol. Cell. Neurosci. 2018, 87, 18–26. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Dupree, J.; Wang, J.; Sandoval, J.; Li, J.; Liu, H.; Shi, Y.; Nave, K.A.; Casaccia-Bonnefil, P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron 2007, 55, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Stolt, C.C.; Rehberg, S.; Ader, M.; Lommes, P.; Riethmacher, D.; Schachner, M.; Bartsch, U.; Wegner, M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor SOX10. Genes Dev. 2002, 16, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, M.; Jaggi, S.; Enz, L.S.; Jacob, C.; Schaeren-Wiemers, N. Theophylline induces remyelination and functional recovery in a mouse model of peripheral neuropathy. Biomedicines 2022, 10, 1418. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Sandoval, J.; Swiss, V.A.; Li, J.; Dupree, J.; Franklin, R.J.; Casaccia-Bonnefil, P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 2008, 11, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, B.; Segel, M.; Chalut, K.J.; Franklin, R.J.M. Remyelination and ageing: Reversing the ravages of time. Mult. Scler. 2019, 25, 1835–1841. [Google Scholar] [CrossRef] [Green Version]
- Graves, J.S.; Krysko, K.M.; Hua, L.H.; Absinta, M.; Franklin, R.J.; Segal, B.M. Ageing and multiple sclerosis. Lancet Neurol. 2023, 22, 66–77. [Google Scholar] [CrossRef]
- Huang, J.K.; Jarjour, A.A.; Nait Oumesmar, B.; Kerninon, C.; Williams, A.; Krezel, W.; Kagechika, H.; Bauer, J.; Zhao, C.; Evercooren, A.B.-V.; et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat. Neurosci. 2010, 14, 45–53. [Google Scholar] [CrossRef] [Green Version]
- McMurran, C.E.; Mukherjee, T.; Brown, J.W.; Michell, A.W.; Chard, D.T.; Franklin, R.J.; Coles, A.J.; Cunniffe, N.G. Remyelination in humans due to a retinoid-X receptor agonist is age-dependent. Ann. Clin. Transl. Neurol. 2022, 9, 1090–1094. [Google Scholar] [CrossRef]
- Shields, S.A.; Gilson, J.M.; Blakemore, W.F.; Franklin, R.J.M. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia 1999, 28, 77–83. [Google Scholar] [CrossRef]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell Stem Cell 2019, 25, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-R.; Zhu, X.; Xiao, Y.; Gu, H.-M.; Zheng, S.-S.; Li, L.; Wang, F.; Dong, Z.-J.; Wang, D.-X.; Wu, Y.; et al. Restoring nuclear entry of SIRTUIN 2 in oligodendrocyte progenitor cells promotes remyelination during ageing. Nat. Commun. 2022, 13, 1225. [Google Scholar] [CrossRef]
- Wu, Y.; Xiong, J.; He, Z.; Yuan, Y.; Wang, B.; Xu, J.; Wu, M.; Zhang, S.; Cai, S.; Zhao, J.; et al. Metformin promotes microglial cells to facilitate myelin debris clearance and accelerate nerve repairment after spinal cord injury. Acta Pharmacol. Sin. 2021, 43, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Segel, M.; Neumann, B.; Hill, M.F.; Weber, I.P.; Viscomi, C.; Zhao, C.; Young, A.; Agley, C.C.; Thompson, A.J.; Gonzalez, G.A.; et al. Niche stiffness underlies the ageing of central nervous system progenitor cells. Nature 2019, 573, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, F.; Fancy, S.P.; Shen, Y.-A.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for Therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordano, C.; Sin, J.H.; Timmons, G.; Yiu, H.H.; Stebbins, K.; Guglielmetti, C.; Cruz-Herranz, A.; Xin, W.; Lorrain, D.; Chan, J.R.; et al. Validating visual evoked potentials as a preclinical, quantitative biomarker for remyelination efficacy. Brain 2022, 145, 3943–3952. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (REBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef] [Green Version]
- Abdelhak, A.; Cordano, C.; Boscardin, W.J.; Caverzasi, E.; Kuhle, J.; Chan, B.; Gelfand, J.M.; Yiu, H.H.; Oertel, F.C.; Beaudry-Richard, A.; et al. Plasma neurofilament light chain levels suggest neuroaxonal stability following therapeutic remyelination in people with multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 972–977. [Google Scholar] [CrossRef]
- Sedel, F.; Bernard, D.; Mock, D.M.; Tourbah, A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology 2016, 110, 644–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cree, B.A.; Cutter, G.; Wolinsky, J.S.; Freedman, M.S.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Arnold, D.; Kuhle, J.; Block, V.; et al. Safety and efficacy of MD1003 (high-dose biotin) in patients with progressive multiple sclerosis (SPI2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2020, 19, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Shao, Z.; Ji, B.; Wang, Q.; Meng, G.; Walus, L.; Lee, X.; Hu, Y.; Graff, C.; Garber, E.; et al. Exposure levels of Anti-LINGO-1 LI81 antibody in the central nervous system and dose-efficacy relationships in rat spinal cord remyelination models after Systemic Administration. J. Pharmacol. Exp. Ther. 2011, 339, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, J.Q.; Rana, J.; Barkhof, F.; Melamed, I.; Gevorkyan, H.; Wattjes, M.P.; de Jong, R.; Brosofsky, K.; Ray, S.; Xu, L.; et al. Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e18. [Google Scholar] [CrossRef] [Green Version]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Frederiksen, J.; Skeen, M.; Jaffe, G.J.; Butzkueven, H.; et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 189–199. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Fischer, B.; Norra, C.; Schellenberger, F.; Stender, N.; Stiefel, M.; Sirén, A.-L.; Paulus, W.; Nave, K.-A.; Gold, R.; et al. Exploring recombinant human erythropoietin in Chronic Progressive Multiple Sclerosis. Brain 2007, 130, 2577–2588. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, K.; Magyari, M.; Sellebjerg, F.; Iversen, P.; Garde, E.; Madsen, C.G.; Börnsen, L.; Romme Christensen, J.; Ratzer, R.; Siebner, H.R.; et al. High-dose erythropoietin in patients with progressive multiple sclerosis: A randomized, placebo-controlled, phase 2 trial. Mult. Scler. J. 2016, 23, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Kato, H.; Yamashita, S.; Aramaki, S.; Morioka, H.; Koresawa, M.; Miyauchi, F.; Toyoshima, H.; Torigoe, T. Effects of domperidone on serum prolactin levels in human beings. Endocrinol. Jpn. 1980, 27, 521–525. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.W.; Sage, K.; Kaur, S.; Kim, J.; Cerchiaro, G.; Yong, V.W.; Cutter, G.R.; Metz, L.M. Repurposing domperidone in secondary progressive multiple sclerosis. Neurology 2021, 96, e2313–e2322. [Google Scholar] [CrossRef]
- Xu, H.; Yang, H.-J.; Li, X.-M. Differential effects of antipsychotics on the development of rat oligodendrocyte precursor cells exposed to cuprizone. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 264, 121–129. [Google Scholar] [CrossRef]
- Watzlawik, J.; Holicky, E.; Edberg, D.D.; Marks, D.L.; Warrington, A.E.; Wright, B.R.; Pagano, R.E.; Rodriguez, M. Human remyelination promoting antibody inhibits apoptotic signaling and differentiation through Lyn kinase in primary rat oligodendrocytes. Glia 2010, 58, 1782–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, B.M.; Bowen, J.D.; Alvarez, E.; Rodriguez, M.; Caggiano, A.O.; Warrington, A.E.; Zhao, P.; Eisen, A. A double-blind, placebo-controlled, single-ascending-dose intravenous infusion study of rhigm22 in subjects with multiple sclerosis immediately following a relapse. Mult. Scler. J. Exp. Trans. Clin. 2022, 8, 205521732210914. [Google Scholar] [CrossRef] [PubMed]
- Magalon, K.; Zimmer, C.; Cayre, M.; Khaldi, J.; Bourbon, C.; Robles, I.; Tardif, G.; Viola, A.; Pruss, R.M.; Bordet, T.; et al. Olesoxime accelerates myelination and promotes repair in models of demyelination. Ann. Neurol. 2012, 71, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Ranjeva, J.; Tourbah, A.; Edan, G.; Barillot, C.; Mer, S.L.; Audoin, B.; Lamy, A.R.; Crespy, L.; Ridley, B.G.; et al. Results of a Phase 1b study to confirm safety and tolerability of olesoxime in multiple sclerosis patients (P7.282). Neurology 2015, 84, P7.282. [Google Scholar]
- Gregg, C.; Shikar, V.; Larsen, P.; Mak, G.; Chojnacki, A.; Yong, V.W.; Weiss, S. White matter plasticity and enhanced remyelination in the maternal CNS. J. Neurosci. 2007, 27, 1812–1823. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Compound | Mechanism of Action—Preclinical | Phase 1 | Phase 2 | Phase 3 |
|---|---|---|---|---|
| Clemastine | Antimuscarinic [107] | VEP100 latency reduced in treated group [109] | Plasma NF light-chain lower in treated group [110] | Recruiting (NCT05338450) |
| Biotin | Improves fatty acid synthesis via cofactors ACC1/2 [111] | Improved motor and visual function [111] | No significant improvement in walking [112] | |
| Bexarotene | RXR-gamma agonist [99] | Improved VEP latency only in patients up to 43 years old [100] | ||
| Opicinumab | Blocks LINGO-1 signaling [113] | Safe and well tolerated [114] | No significant remyelination [115] | |
| Erythopoietin | Prevents brain atrophy [116] | Improved motor function in treated group [116] | No clinical significance [117] | |
| Domperidone | Promotes prolactin secretion [118] | Did not slow disease progression [119] | ||
| Quetiapine | Antioxidative and pro-proliferative to OPCs [120] | Completed but not published (NCT02087631) | ||
| rHIgM22 | Inhibits apoptosis via Lyn kinase [121] | Safe and well tolerated [122] | ||
| Benztropine | Downregulates Notch1 [106] | |||
| Metformin | Rejuvenates OPCs and improves autophagy [102] | Recruiting (NCT05349474, NCT04121468, NCT05131828, NCT05298670) | ||
| Theophylline | Activates HDAC2, increases Sox10 [75] | |||
| Miconazole | Phosphorylates ERK1/2 [80] | |||
| Olesoxime | Directly stimulates OPC maturation [123] | Safe as an add-on therapy to interferon-beta [124] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Jacob, C. Mechanisms of Demyelination and Remyelination Strategies for Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 6373. https://doi.org/10.3390/ijms24076373
Zhao X, Jacob C. Mechanisms of Demyelination and Remyelination Strategies for Multiple Sclerosis. International Journal of Molecular Sciences. 2023; 24(7):6373. https://doi.org/10.3390/ijms24076373
Chicago/Turabian StyleZhao, Xinda, and Claire Jacob. 2023. "Mechanisms of Demyelination and Remyelination Strategies for Multiple Sclerosis" International Journal of Molecular Sciences 24, no. 7: 6373. https://doi.org/10.3390/ijms24076373