
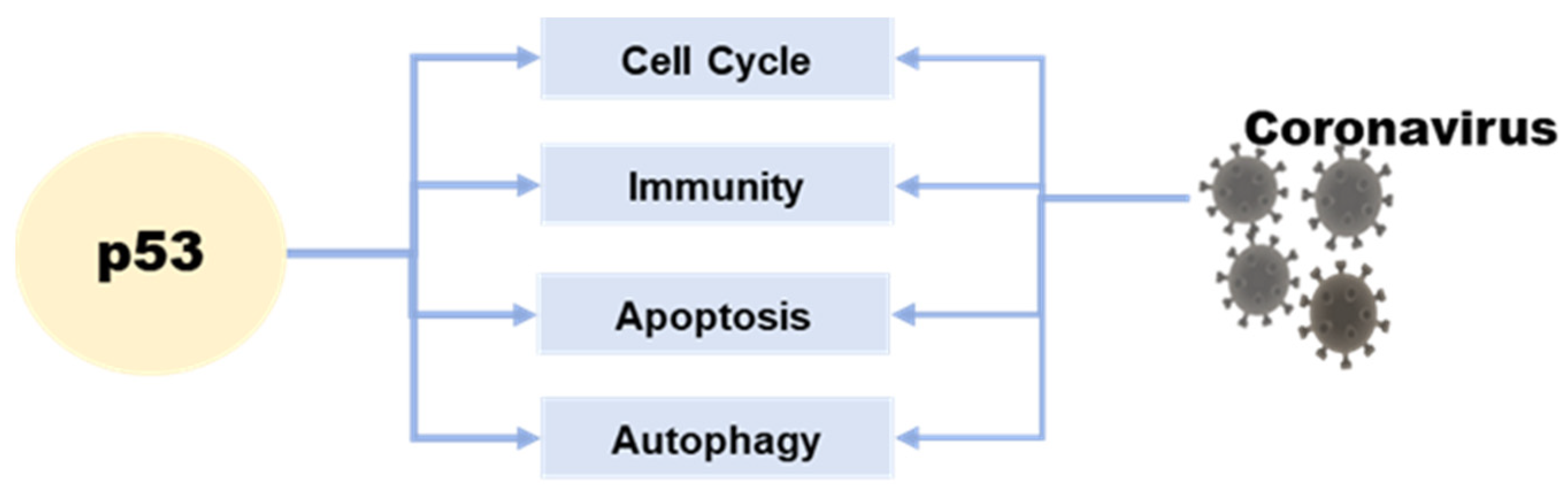
Roles of p53-Mediated Host–Virus Interaction in Coronavirus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
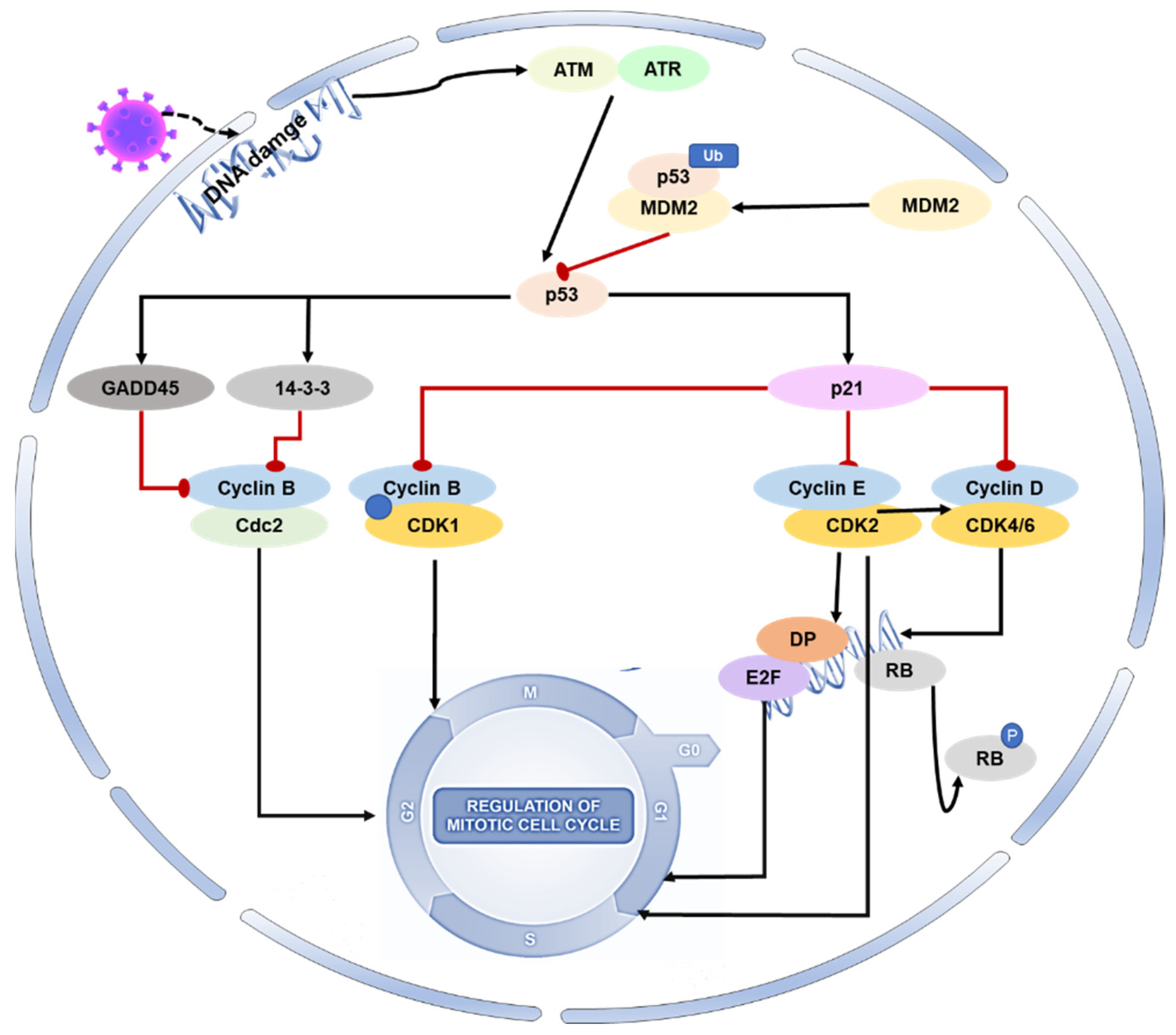
2. p53 Affects Coronaviruses by Regulating the Cell Cycle
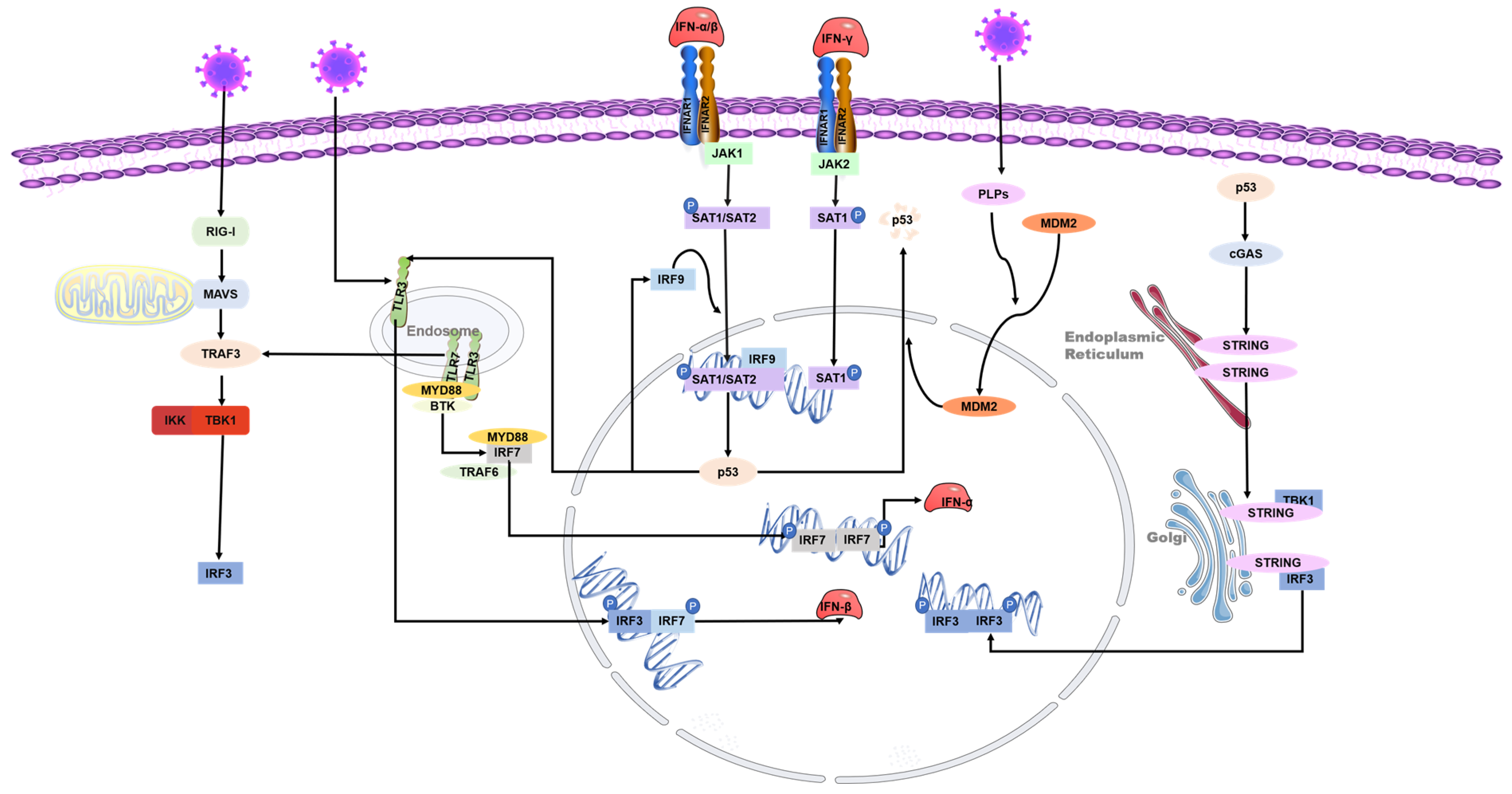
3. The Interaction between p53 and Interferon
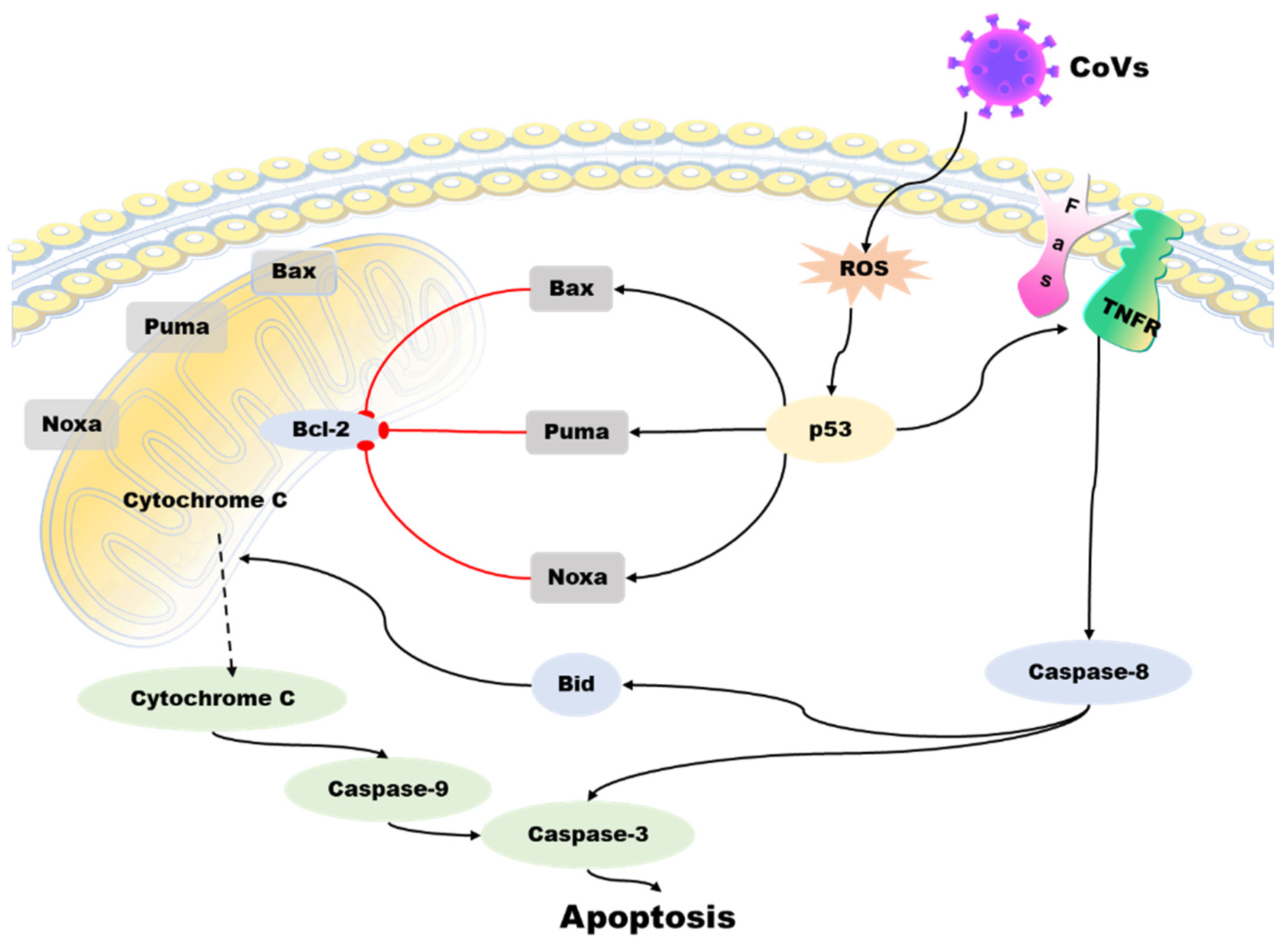
4. p53 Affects Coronavirus-Associated Apoptosis
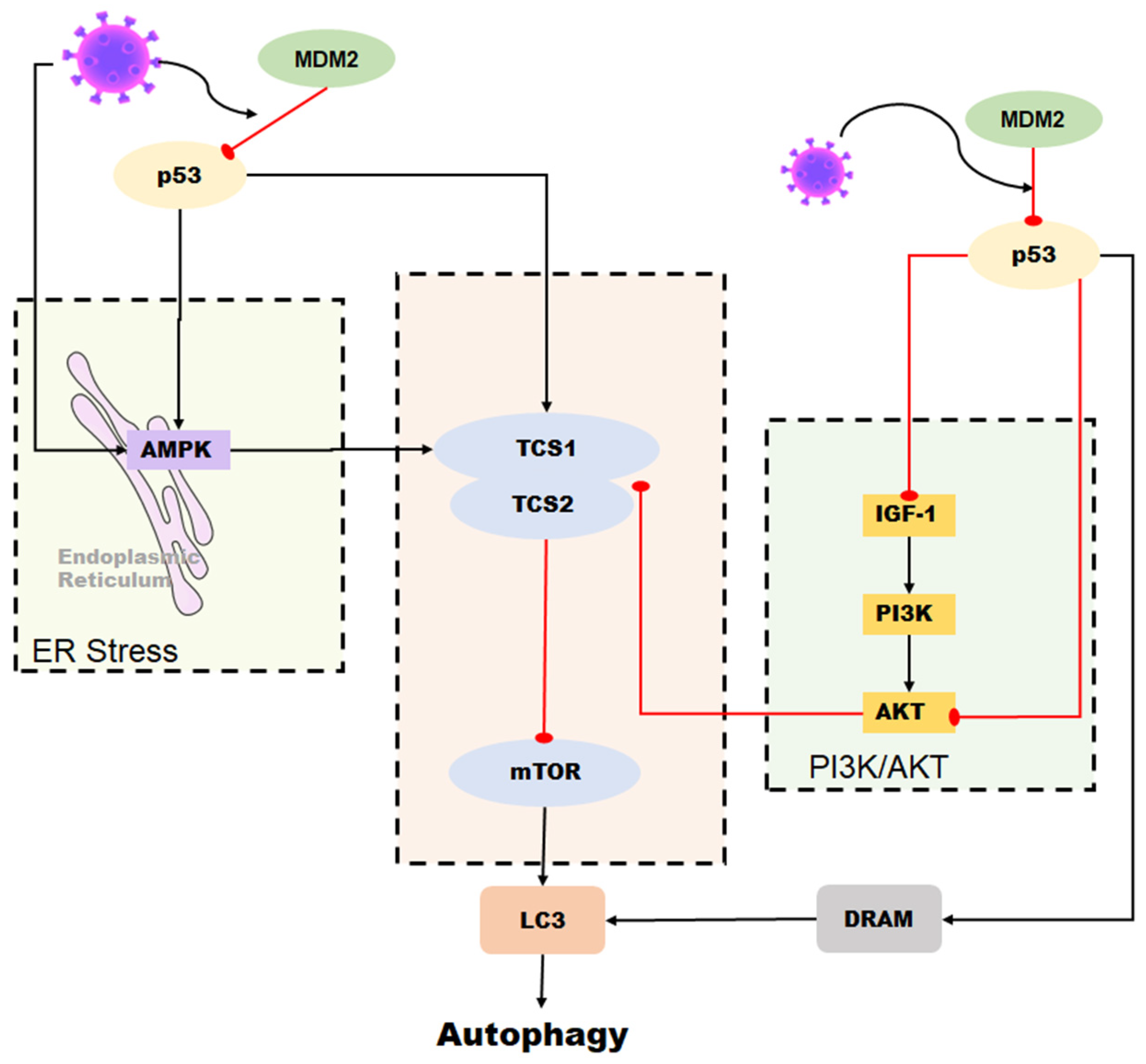
5. p53 Affects Coronavirus-Associated Autophagy
6. Coronavirus Proteins Interact with p53
7. p53 Is a Potential Target for Anti-Coronavirus Drugs
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Enjuanes, L.; Almazán, F.; Sola, I.; Zuñiga, S. Biochemical aspects of coronavirus replication and virus-host interaction. Annu. Rev. Microbiol. 2006, 60, 211–230. [Google Scholar] [CrossRef] [Green Version]
- Amarilla, A.A.; Sng, J.D.J.; Parry, R.; Deerain, J.M.; Potter, J.R.; Setoh, Y.X.; Rawle, D.J.; Le, T.T.; Modhiran, N.; Wang, X.; et al. A versatile reverse genetics platform for SARS-CoV-2 and other positive-strand RNA viruses. Nat. Commun. 2021, 12, 3431. [Google Scholar] [CrossRef]
- Wang, L.-F.; Shi, Z.; Zhang, S.; Field, H.; Daszak, P.; Eaton, B.T. Review of bats and SARS. Emerg. Infect. Dis. 2006, 12, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Enserink, M. Infectious diseases. Calling all coronavirologists. Science 2003, 300, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Sui, L.; Li, L.; Zhao, Y.; Zhao, Y.; Hao, P.; Guo, X.; Wang, W.; Wang, G.; Li, C.; Liu, Q. Host cell cycle checkpoint as antiviral target for SARS-CoV-2 revealed by integrative transcriptome and proteome analyses. Signal Transduct. Target. Ther. 2023, 8, 21. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; Robertson, D.L. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Wang, Z.; Gutiérrez-Castrellón, P.; Shi, H. Cell deaths: Involvement in the pathogenesis and intervention therapy of COVID-19. Signal Transduct. Target. Ther. 2022, 7, 186. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Vousden, K.H. p53: New roles in metabolism. Trends Cell Biol. 2007, 17, 286–291. [Google Scholar] [CrossRef]
- Levine, B.; Abrams, J. p53: The Janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, D.; Rotter, V. p53-dependent cell cycle control: Response to genotoxic stress. Semin. Cancer Biol. 1998, 8, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Offer, H.; Zurer, I.; Banfalvi, G.; Reha’k, M.; Falcovitz, A.; Milyavsky, M.; Goldfinger, N.; Rotter, V. p53 modulates base excision repair activity in a cell cycle-specific manner after genotoxic stress. Cancer Res. 2001, 61, 88–96. [Google Scholar]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmueli, A.; Oren, M. Regulation of p53 by Mdm2: Fate is in the numbers. Mol. Cell 2004, 13, 4–5. [Google Scholar] [CrossRef]
- Guo, G.; Cui, Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J. Immunother. Cancer 2015, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Malumbres, M.; Sicinski, P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Seo, H.R.; Lee, D.H.; Lee, H.J.; Baek, M.; Bae, S.; Soh, J.W.; Lee, S.J.; Kim, J.; Lee, Y.S. Cyclin G1 overcomes radiation-induced G2 arrest and increases cell death through transcriptional activation of cyclin B1. Cell Death Differ. 2006, 13, 1475–1484. [Google Scholar] [CrossRef] [Green Version]
- Braithwaite, A.W.; Prives, C.L. p53: More research and more questions. Cell Death Differ. 2006, 13, 877–880. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Grams, T.R.; Bloom, D.C.; Toth, Z. Signaling Pathway Reporter Screen with SARS-CoV-2 Proteins Identifies nsp5 as a Repressor of p53 Activity. Viruses 2022, 14, 1039. [Google Scholar] [CrossRef] [PubMed]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef]
- Chander, Y.; Kumar, R.; Khandelwal, N.; Singh, N.; Shringi, B.N.; Barua, S.; Kumar, N. Role of p38 mitogen-activated protein kinase signalling in virus replication and potential for developing broad spectrum antiviral drugs. Rev. Med. Virol. 2021, 31, 1–16. [Google Scholar] [CrossRef]
- Kyriakopoulos, A.M.; Nigh, G.; McCullough, P.A.; Seneff, S. Mitogen Activated Protein Kinase (MAPK) Activation, p53, and Autophagy Inhibition Characterize the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Spike Protein Induced Neurotoxicity. Cureus 2022, 14, e32361. [Google Scholar] [CrossRef]
- Ryan, E.L.; Hollingworth, R.; Grand, R.J. Activation of the DNA Damage Response by RNA Viruses. Biomolecules 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chen, J.; Hu, D.; Pan, P.; Liang, L.; Wu, W.; Tang, Y.; Huang, X.R.; Yu, X.; Wu, J.; et al. SARS-CoV-2 N Protein Induces Acute Kidney Injury via Smad3-Dependent G1 Cell Cycle Arrest Mechanism. Adv. Sci. 2022, 9, e2103248. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Shi, D.; Xing, X.; Qi, S.; Yang, D.; Zhang, J.; Han, Y.; Zhu, Q.; Sun, H.; Wang, X.; et al. Coronavirus Porcine Epidemic Diarrhea Virus Nucleocapsid Protein Interacts with p53 to Induce Cell Cycle Arrest in S-Phase and Promotes Viral Replication. J. Virol. 2021, 95, e0018721. [Google Scholar] [CrossRef]
- Chen, C.-J.; Makino, S. Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 2004, 78, 5658–5669. [Google Scholar] [CrossRef] [Green Version]
- Dove, B.; Brooks, G.; Bicknell, K.; Wurm, T.; Hiscox, J.A. Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 2006, 80, 4147–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surjit, M.; Liu, B.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 2006, 281, 10669–10681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Wu, H.; Huang, J.; Xu, Y.; Yang, F.; Zhang, Q.; Xu, X. Porcine epidemic diarrhea virus through p53-dependent pathway causes cell cycle arrest in the G0/G1 phase. Virus Res. 2018, 253, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Fontela, C.; Macip, S.; Martínez-Sobrido, L.; Brown, L.; Ashour, J.; García-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Wang, X.; Guo, L.; Qiu, Y.; Li, X.; Yu, H.; Xiang, H.; Tong, G.; Ma, Z. Influenza A virus induces p53 accumulation in a biphasic pattern. Biochem. Biophys. Res. Commun. 2009, 382, 331–335. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Response to Viruses for IFN-α/β Gene Induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Saha, S.; Li, J.; Montrose, D.C.; Martinez, L.A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 2023, 83, 266–280.e266. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Poon, L.L.M.; Ng, I.H.Y.; Luk, W.; Sia, S.-F.; Wu, M.H.S.; Chan, K.-H.; Yuen, K.-Y.; Gordon, S.; Guan, Y.; et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: Possible relevance to pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.-N.; You, Y.; Cui, X.-M.; Gao, H.-X.; Wang, G.-L.; Zhang, S.-B.; Yao, L.; Duan, L.-J.; Zhu, K.-L.; Wang, Y.-L.; et al. Single-cell immune profiling reveals distinct immune response in asymptomatic COVID-19 patients. Signal Transduct. Target. Ther. 2021, 6, 342. [Google Scholar] [CrossRef] [PubMed]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Harford, J.B.; Kim, S.S.; Pirollo, K.F.; Chang, E.H. TP53 Gene Therapy as a Potential Treatment for Patients with COVID-19. Viruses 2022, 14, 739. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Müller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Bortot, B.; Romani, A.; Ricci, G.; Biffi, S. Exploiting Extracellular Vesicles Strategies to Modulate Cell Death and Inflammation in COVID-19. Front. Pharmacol. 2022, 13, 877422. [Google Scholar] [CrossRef] [PubMed]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, 369, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Thompson, C.B. Apoptosis and disease: Regulation and clinical relevance of programmed cell death. Annu. Rev. Med. 1997, 48, 267–281. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, M.S.; Fornace, A.J. Death and decoy receptors and p53-mediated apoptosis. Leukemia 2000, 14, 1509–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 FAMILY: Regulators of Cell Death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. Living with p53, Dying of p53. Cell 2007, 130, 597–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Xu, Y.; Zhang, Q.; Yang, F.; Yin, Z.; Wang, L.; Li, Q. Porcine epidemic diarrhea virus infections induce apoptosis in Vero cells via a reactive oxygen species (ROS)/p53, but not p38 MAPK and SAPK/JNK signalling pathways. Vet. Microbiol. 2019, 232, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, A.; Wu, D.; Wang, C.; Huang, M.; Xiong, X.; Jin, L.; Zhou, W.; Qiu, Y.; Zhou, X. Dual inhibition of innate immunity and apoptosis by human cytomegalovirus protein UL37x1 enables efficient virus replication. Nat. Microbiol. 2022, 7, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Slonchak, A.; Hugo, L.E.; Freney, M.E.; Hall-Mendelin, S.; Amarilla, A.A.; Torres, F.J.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Hall, R.A.; et al. Zika virus noncoding RNA suppresses apoptosis and is required for virus transmission by mosquitoes. Nat. Commun. 2020, 11, 2205. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, S.; Saeed, M.; Giorgi, C.; Li, J.; Hoffmann, H.H.; Pinton, P.; Rice, C.M.; Pagano, M. NS5A Promotes Constitutive Degradation of IP3R3 to Counteract Apoptosis Induced by Hepatitis C Virus. Cell Rep. 2018, 25, 833–840.e833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, C. Porcine deltacoronavirus induces caspase-dependent apoptosis through activation of the cytochrome c-mediated intrinsic mitochondrial pathway. Virus Res. 2018, 253, 112–123. [Google Scholar] [CrossRef]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-level inhibition of coronavirus replication by chemical ER stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef]
- Lodi, G.; Gentili, V.; Casciano, F.; Romani, A.; Zauli, G.; Secchiero, P.; Zauli, E.; Simioni, C.; Beltrami, S.; Fernandez, M.; et al. Cell cycle block by p53 activation reduces SARS-CoV-2 release in infected alveolar basal epithelial A549-hACE2 cells. Front. Pharmacol. 2022, 13, 1018761. [Google Scholar] [CrossRef]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Rep. 2020, 20, 100765. [Google Scholar] [CrossRef]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Mlcochova, P. Cyclin D3 restricts SARS-CoV-2 envelope incorporation into virions and interferes with viral spread. EMBO J. 2022, 41, e111653. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’Kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid Protein Recruitment to Replication-Transcription Complexes Plays a Crucial Role in Coronaviral Life Cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Draczkowski, P.; Wang, Y.S.; Chang, C.Y.; Chien, Y.C.; Cheng, Y.H.; Wu, Y.M.; Wang, C.H.; Chang, Y.C.; Chang, Y.C.; et al. In Situ structure and dynamics of an alphacoronavirus spike protein by cryo-ET and cryo-EM. Nat. Commun. 2022, 13, 4877. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Huang, Y.; Du, Q.; Dong, F.; Zhao, X.; Zhang, W.; Xu, X.; Tong, D. TGEV nucleocapsid protein induces cell cycle arrest and apoptosis through activation of p53 signaling. Biochem. Biophys. Res. Commun. 2014, 445, 497–503. [Google Scholar] [CrossRef]
- Zhao, H.; Bauzon, F.; Fu, H.; Lu, Z.; Cui, J.; Nakayama, K.; Nakayama, K.I.; Locker, J.; Zhu, L. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 2013, 24, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Sugiyama, K.; Kubo, H.; Huang, C.; Makino, S. Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J. Virol. 2004, 78, 10410–10419. [Google Scholar] [CrossRef] [Green Version]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Burch, C.L.; Chao, L. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature 2000, 406, 625–628. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In Vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, B.; Deng, L.; Liang, B.; Ping, J. Virus-host interaction networks as new antiviral drug targets for IAV and SARS-CoV-2. Emerg. Microbes Infect. 2022, 11, 1371–1389. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.M.; Podyminogin, R.L.; Diercks, A.H.; Treuting, P.M.; Peschon, J.J.; Rodriguez, D.; Gundapuneni, M.; Weiss, M.J.; Aderem, A. miR-144 attenuates the host response to influenza virus by targeting the TRAF6-IRF7 signaling axis. PLoS Pathog. 2017, 13, e1006305. [Google Scholar] [CrossRef] [Green Version]
- Artese, A.; Svicher, V.; Costa, G.; Salpini, R.; Di Maio, V.C.; Alkhatib, M.; Ambrosio, F.A.; Santoro, M.M.; Assaraf, Y.G.; Alcaro, S.; et al. Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2020, 53, 100721. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; de Oliveira, A.P.G. Targeting the Prion-like Aggregation of Mutant p53 to Combat Cancer. Acc. Chem. Res. 2018, 51, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, X.; Wang, G.; Yang, Y.; Yuan, Y.; Ouyang, L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 2019, 176, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Tsurumi, T. Genome guardian p53 and viral infections. Rev. Med. Virol. 2013, 23, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. p53 and the Viral Connection: Back into the Future (‡). Cancers 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Alzhanova, D.; Corcoran, K.; Bailey, A.G.; Long, K.; Taft-Benz, S.; Graham, R.L.; Broussard, G.S.; Heise, M.; Neumann, G.; Halfmann, P.; et al. Novel modulators of p53-signaling encoded by unknown genes of emerging viruses. PLoS Pathog. 2021, 17, e1009033. [Google Scholar] [CrossRef]
- Singh, N.; Bharara Singh, A. S2 subunit of SARS-nCoV-2 interacts with tumor suppressor protein p53 and BRCA: An in silico study. Transl. Oncol. 2020, 13, 100814. [Google Scholar] [CrossRef]
- Yuan, L.; Chen, Z.; Song, S.; Wang, S.; Tian, C.; Xing, G.; Chen, X.; Xiao, Z.X.; He, F.; Zhang, L. p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J. Biol. Chem. 2015, 290, 3172–3182. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Abd El Maksoud, A.I.; Roshdey, T.; El-Masry, S. Guava flavonoid glycosides prevent influenza A virus infection via rescue of P53 activity. J. Med. Virol. 2019, 91, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Song, X.; Li, C.; Hu, H.; Li, W.; Wang, L.; Hu, J.; Liao, C.; Liang, H.; He, Z.; et al. Chrysin Ameliorates Influenza Virus Infection in the Upper Airways by Repressing Virus-Induced Cell Cycle Arrest and Mitochondria-Dependent Apoptosis. Front. Immunol. 2022, 13, 872958. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Liu, Y.; Xie, J.; Hu, C.; Wang, X. Role of p53 in pseudorabies virus replication, pathogenicity, and host immune responses. Vet. Res. 2019, 50, 9. [Google Scholar] [CrossRef] [Green Version]
- Casavant, N.C.; Luo, M.H.; Rosenke, K.; Winegardner, T.; Zurawska, A.; Fortunato, E.A. Potential Role for p53 in the Permissive Life Cycle of Human Cytomegalovirus. J. Virol. 2006, 80, 8390–8401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zauli, G.; AlHilali, S.; Al-Swailem, S.; Secchiero, P.; Voltan, R. Therapeutic potential of the MDM2 inhibitor Nutlin-3 in counteracting SARS-CoV-2 infection of the eye through p53 activation. Front. Med. 2022, 9, 902713. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Wang, J.; Zhang, J.; Duan, C.; Wang, J. Ergosterol Peroxide Inhibits Porcine Epidemic Diarrhea Virus Infection in Vero Cells by Suppressing ROS Generation and p53 Activation. Viruses 2022, 14, 402. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; Li, K.; Yang, M.; Wang, Q.; Hao, Z. Triacetyl Resveratrol Inhibits PEDV by Inducing the Early Apoptosis In Vitro. Int. J. Mol. Sci. 2022, 23, 14499. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Liu, Y.; Li, K.; Hao, Z. Roles of p53-Mediated Host–Virus Interaction in Coronavirus Infection. Int. J. Mol. Sci. 2023, 24, 6371. https://doi.org/10.3390/ijms24076371
Wang X, Liu Y, Li K, Hao Z. Roles of p53-Mediated Host–Virus Interaction in Coronavirus Infection. International Journal of Molecular Sciences. 2023; 24(7):6371. https://doi.org/10.3390/ijms24076371
Chicago/Turabian StyleWang, Xue, Yi Liu, Kaiyuan Li, and Zhihui Hao. 2023. "Roles of p53-Mediated Host–Virus Interaction in Coronavirus Infection" International Journal of Molecular Sciences 24, no. 7: 6371. https://doi.org/10.3390/ijms24076371