
Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders
 , , and
, , and
Abstract
:1. Introduction
2. Neurotrophic Factors in the Healthy and Diseased Brain
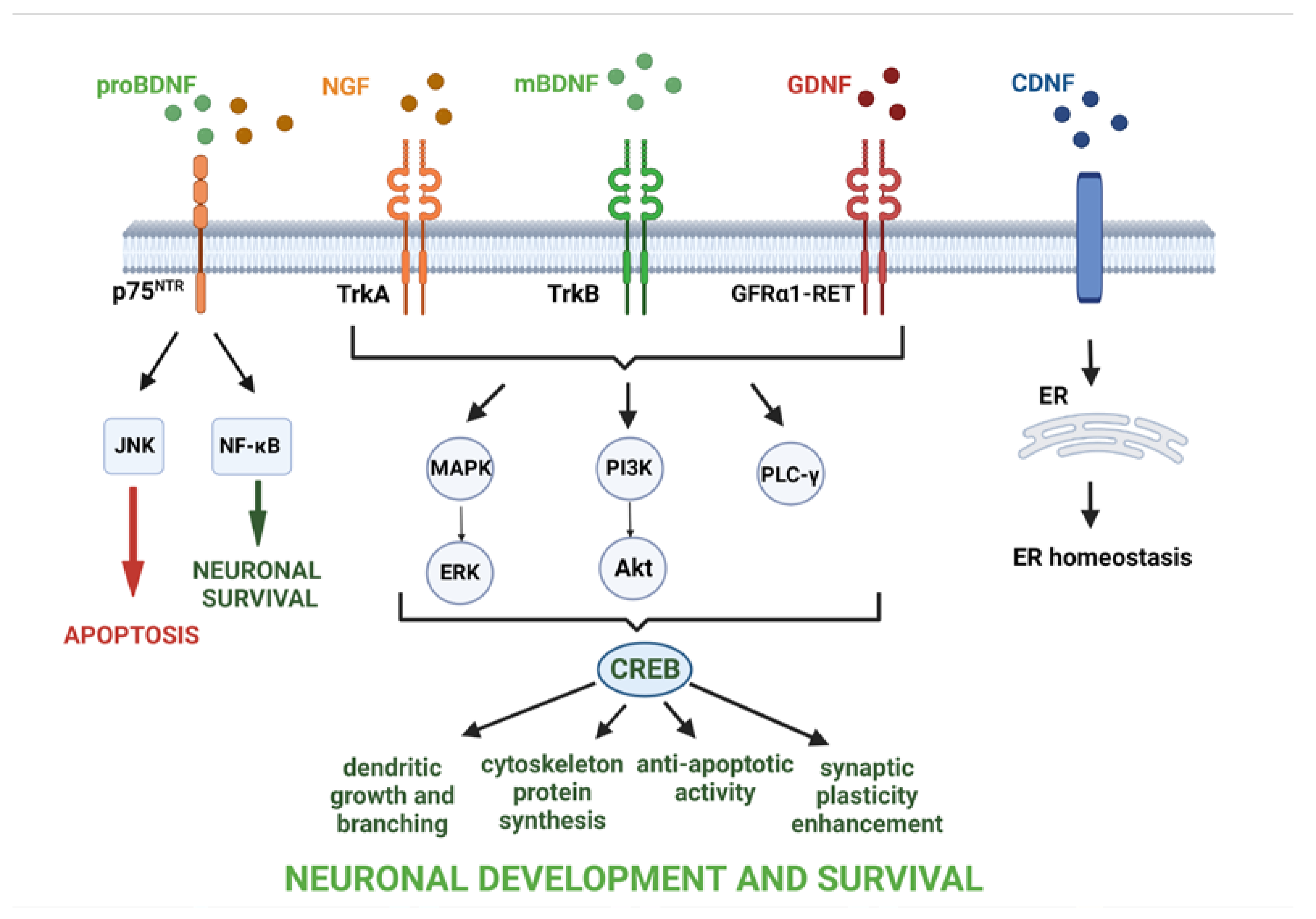
2.1. BDNF
2.2. GDNF
2.3. CDNF
2.4. NGF
3. Neurotrophic Factors in Glia-Neuronal Crosstalk and Their Role in Neuroinflammation
3.1. The Role of BDNF in Neuroinflammation
3.2. The Role of GDNF in Neuroinflammation
3.3. The Role of CDNF in Neuroinflammation
3.4. The Role of NGF in Neuroinflammation
4. Possible Interaction between Neurotrophic Factors, Mitochondria, and Neuroinflammation
5. Conclusions and Further Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DiSabato, D.; Quan, N.; Godbout, J.P. The Devil Is in the Details. J. Neurochem. 2017, 139, 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, A.M.; Rodríguez, J.; Giambartolomei, G.H. Microglia at the Crossroads of Pathogen-Induced Neuroinflammation. ASN Neuro 2022, 14, 175909142211045. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H. Roles of Glial Cells in Neuroinflammation and Neurodegeneration. Clin. Exp. Neuroimmunol. 2013, 4, 2–16. [Google Scholar] [CrossRef]
- Jha, M.K.; Jeon, S.; Suk, K. Glia as a Link between Neuroinflammation and Neuropathic Pain. Immune Netw. 2012, 12, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
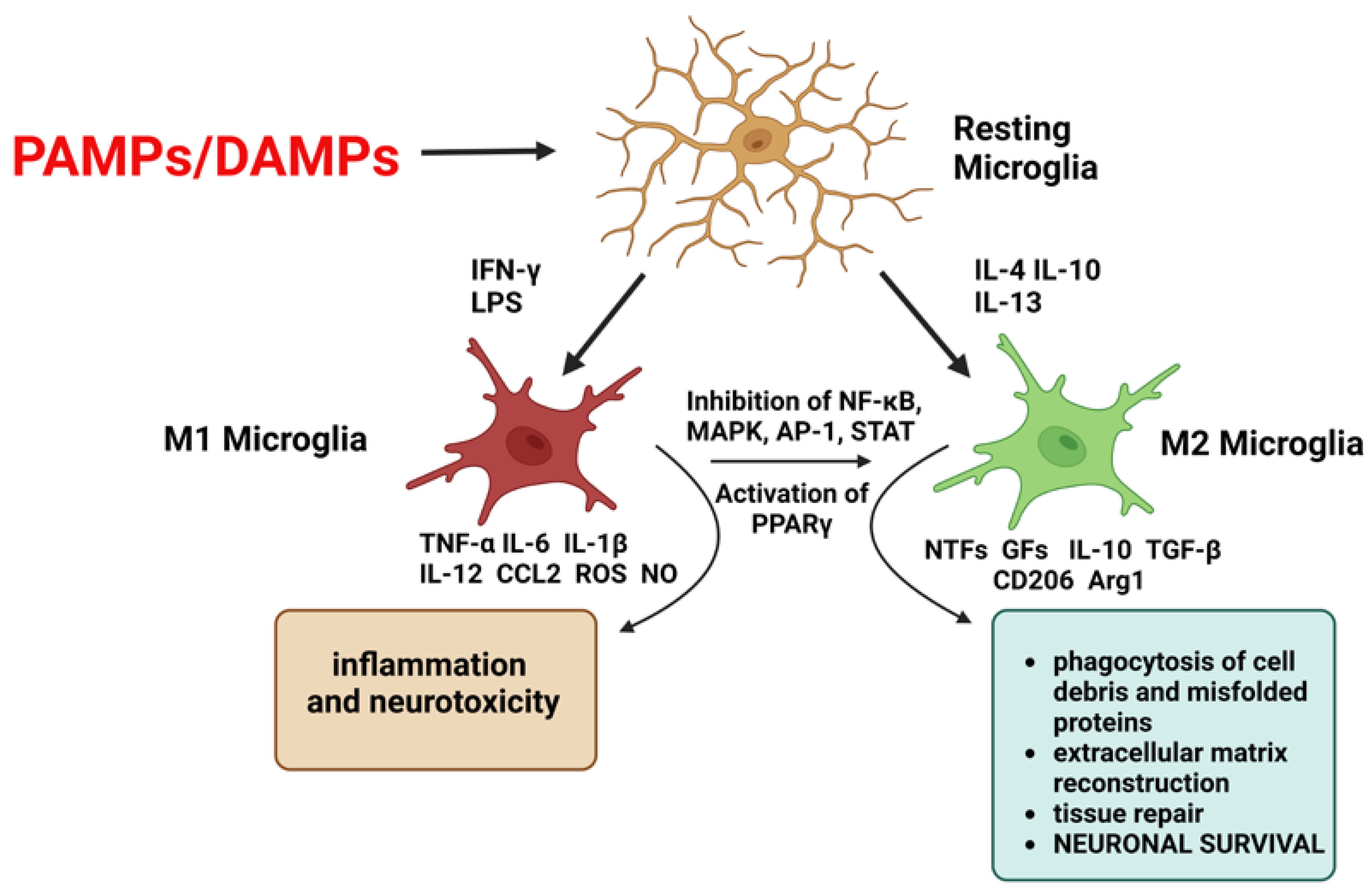
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14. [Google Scholar] [CrossRef]
- Liu, X.; Ma, J.; Ding, G.; Gong, Q.; Wang, Y.; Yu, H.; Cheng, X. Microglia Polarization from M1 toward M2 Phenotype Is Promoted by Astragalus Polysaccharides Mediated through Inhibition of MiR-155 in Experimental Autoimmune Encephalomyelitis. Oxidative Med. Cell. Longev. 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Qie, S.; Ran, Y.; Lu, X.; Su, W.; Li, W.; Xi, J.; Gong, W.; Liu, Z. Candesartan Modulates Microglia Activation and Polarization via NF-ΚB Signaling Pathway. Int. J. Immunopathol. Pharmacol. 2020, 34, 205873842097490. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, Y.-Z.; Wang, G.-Q.; Li, D.-D.; Shi, J.-S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Kang, J.; Liu, S.; Xu, Y.; Shao, B. The Protective Effects of Peroxisome Proliferator-Activated Receptor Gamma in Cerebral Ischemia-Reperfusion Injury. Front. Neurol. 2020, 11, 588516. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Fatoba, O.; Itokazu, T.; Yamashita, T. Microglia as Therapeutic Target in Central Nervous System Disorders. J. Pharmacol. Sci. 2020, 144, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
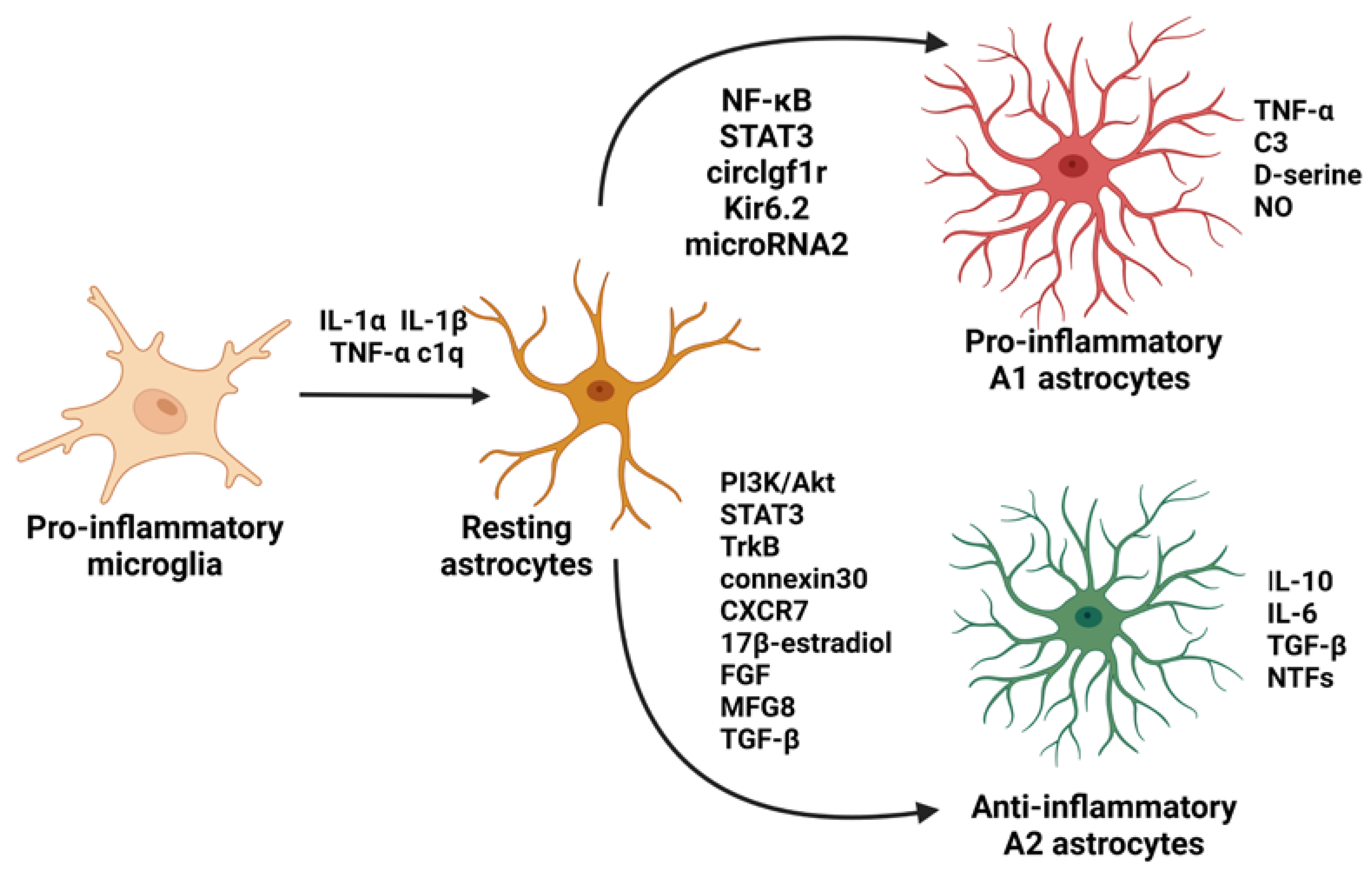
- Verkhratsky, A.; Rodríguez, J.J.; Parpura, V. Astroglia in Neurological Diseases. Future Neurol. 2013, 8, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phatnani, H.; Maniatis, T. Astrocytes in Neurodegenerative Disease: Table 1. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [Green Version]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, S.; Peterson, T.C.; et al. Neurotoxic Reactinve Astroctes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Meirelles, L.; Simon, D.; Regner, A. Neurotrauma: The Crosstalk between Neurotrophins and Inflammation in the Acutely Injured Brain. Int. J. Mol. Sci. 2017, 18, 1082. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.-Y.; Huo, J. A1/A2 Astrocytes in Central Nervous System Injuries and Diseases: Angels or Devils? Neurochem. Int. 2021, 148, 105080. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Jasper, H.; Neves, J. Trophic Factors in Inflammation and Regeneration: The Role of MANF and CDNF. Front. Physiol. 2018, 9, 1629. [Google Scholar] [CrossRef] [Green Version]
- Martorana, F.; Gaglio, D.; Bianco, M.R.; Aprea, F.; Virtuoso, A.; Bonanomi, M.; Alberghina, L.; Papa, M.; Colangelo, A.M. Differentiation by Nerve Growth Factor (NGF) Involves Mechanisms of Crosstalk between Energy Homeostasis and Mitochondrial Remodeling. Cell Death Dis. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Cameron, I.; Franklin, P.; Spedding, M. BDNF Increases Rat Brain Mitochondrial Respiratory Coupling at Complex I, but Not Complex II. Eur. J. Neurosci. 2004, 20, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.J.O.; Bromberg, E.; de Vries, E.F.J. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [Green Version]
- Pałasz, E.; Bąk, A.; Gąsiorowska, A.; Niewiadomska, G. The Role of Trophic Factors and Inflammatory Processes in Physical Activity-Induced Neuroprotection in Parkinson’s Disease. Postepy Hig. Med. Dosw. 2017, 71, 713–726. [Google Scholar] [CrossRef]
- Pöyhönen, S.; Er, S.; Domanskyi, A.; Airavaara, M. Effects of Neurotrophic Factors in Glial Cells in the Central Nervous System: Expression and Properties in Neurodegeneration and Injury. Front. Physiol. 2019, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Thoenen, H. Neurotrophins and Neuronal Plasticity. Science 1995, 270, 593–598. [Google Scholar] [CrossRef]
- Airavaara, M.; Chiocco, M.J.; Howard, D.B.; Zuchowski, K.L.; Peränen, J.; Liu, C.; Fang, S.; Hoffer, B.J.; Wang, Y.; Harvey, B.K. Widespread Cortical Expression of MANF by AAV Serotype 7: Localization and Protection against Ischemic Brain Injury. Exp. Neurol. 2010, 225, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Barcia, C. Glial-Mediated Inflammation Underlying Parkinsonism. Scientifica (Cairo) 2013, 2013, 357805. [Google Scholar] [CrossRef] [Green Version]
- Bath, K.G.; Lee, F.S. Neurotrophic Factor Control of Adult SVZ Neurogenesis. Dev. Neurobiol. 2010, 70, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Beavers, K.M.; Hsu, F.-C.; Isom, S.; Kritchevsky, S.B.; Church, T.; Goodpaster, B.; Pahor, M.; Nickolas, B.J. Long-Term Physical Activity and Inflammatory Biomarkers in Older Adults. Med. Sci. Sport. Exerc. 2010, 42, 2189–2196. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Al-Qudah, M.; Al-Dwairi, A. Mechanisms and Regulation of Neurotrophin Synthesis and Secretion. Neurosciences 2016, 21, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Nickl-Jockschat, T. Neurotrophic Factors in Autism Spectrum Disorders. In Comprehensive Guide to Autism; Springer: New York, NY, USA, 2014; pp. 741–754. [Google Scholar]
- Brambilla, R. Neuroinflammation, the Thread Connecting Neurological Disease. Acta Neuropathol. 2019, 137, 689–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Cieślik, M.; Gąssowska-Dobrowolska, M.; Jęśko, H.; Czapski, G.A.; Wilkaniec, A.; Zawadzka, A.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Boguszewski, P.M.; et al. Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring. Int. J. Mol. Sci. 2020, 21, 4097. [Google Scholar] [CrossRef] [PubMed]
- Paraschivescu, C.; Barbosa, S.; Lorivel, T.; Glaichenhaus, N.; Davidovic, L. Cytokine Changes Associated with the Maternal Immune Activation (MIA) Model of Autism: A Penalized Regression Approach. PLoS ONE 2020, 15, e0231609. [Google Scholar] [CrossRef] [PubMed]
- Orefice, L.L.; Waterhouse, E.G.; Partridge, J.G.; Lalchandani, R.R.; Vicini, S.; Xu, B. Distinct Roles for Somatically and Dendritically Synthesized Brain-Derived Neurotrophic Factor in Morphogenesis of Dendritic Spines. J. Neurosci. 2013, 33, 11618–11632. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Fernandez, J.R.; Zegarek, G.F.; Lo, S.B.; Firestein, B.L. BDNF-Promoted Increases in Proximal Dendrites Occur via CREB-Dependent Transcriptional Regulation of Cypin. J. Neurosci. 2011, 31, 9735–9745. [Google Scholar] [CrossRef] [Green Version]
- Gorski, J.A.; Zeiler, S.R.; Tamowski, S.; Jones, K.R. Brain-Derived Neurotrophic Factor Is Required for the Maintenance of Cortical Dendrites. J. Neurosci. 2003, 23, 6856–6865. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wu, Y.; Wang, Y.; Zhu, J.; Chu, H.; Kong, L.; Yin, L.; Ma, H. Brain-Derived Neurotrophic Factor Increases Synaptic Protein Levels via the MAPK/Erk Signaling Pathway and Nrf2/Trx Axis Following the Transplantation of Neural Stem Cells in a Rat Model of Traumatic Brain Injury. Neurochem. Res. 2017, 42, 3073–3083. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic Localization of PSD-95 Is Regulated by All Three Pathways Downstream of TrkB Signaling. Front. Synaptic. Neurosci. 2014, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, J.C.; Xia, C.-M.; Liu, M.; Shen, S.; Yu, S.J.; Chung, C.; Qiao, L.-Y. Endogenous PI3K/Akt and NMDAR Act Independently in the Regulation of CREB Activity in Lumbosacral Spinal Cord in Cystitis. Exp. Neurol. 2013, 250, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opazo, P.; Watabe, A.M.; Grant, S.G.N.; O’Dell, T.J. Phosphatidylinositol 3-Kinase Regulates the Induction of Long-Term Potentiation through Extracellular Signal-Related Kinase-Independent Mechanisms. J. Neurosci. 2003, 23, 3679–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-H.; Chen, C.-C.; Hung, T.-H.; Chuang, Y.-C.; Chao, M.; Shyue, S.-K.; Chen, S.-F. Activation of TrkB/Akt Signaling by a TrkB Receptor Agonist Improves Long-Term Histological and Functional Outcomes in Experimental Intracerebral Hemorrhage. J. Biomed. Sci. 2019, 26, 53. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Xiong, L.-J.; Tong, Y.; Mao, M. Neuroprotective Effect of Brain-Derived Neurotrophic Factor Mediated by Autophagy through the PI3K/Akt/MTOR Pathway. Mol. Med. Rep. 2013, 8, 1011–1016. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.v.; Krimm, R.F. BDNF Is Required for the Survival of Differentiated Geniculate Ganglion Neurons. Dev. Biol. 2010, 340, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Petersén, Å.; Larsen, K.E.; Behr, G.G.; Romero, N.; Przedborski, S.; Brundin, P.; Sulzer, D. Brain-Derived Neurotrophic Factor Inhibits Apoptosis and Dopamine-Induced Free Radical Production in Striatal Neurons but Does Not Prevent Cell Death. Brain Res. Bull 2001, 56, 331–335. [Google Scholar] [CrossRef]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.F.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF MRNA Expression in the Parkinson’s Disease Substantia Nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef]
- Skogstrand, K.; Hagen, C.M.; Borbye-Lorenzen, N.; Christiansen, M.; Bybjerg-Grauholm, J.; Bækvad-Hansen, M.; Werge, T.; Børglum, A.; Mors, O.; Nordentoft, M.; et al. Reduced Neonatal Brain-Derived Neurotrophic Factor Is Associated with Autism Spectrum Disorders. Transl. Psychiatry 2019, 9, 252. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-H.; Shi, X.-J.; Fan, F.-C.; Cheng, Y. Peripheral Blood Neurotrophic Factor Levels in Children with Autism Spectrum Disorder: A Meta-Analysis. Sci. Rep. 2021, 11, 15. [Google Scholar] [CrossRef]
- Anghelescu, I.; Dettling, M. Neuron Number in Children With Autism. JAMA 2012, 307. [Google Scholar] [CrossRef] [PubMed]
- Lainhart, J.E.; Lange, N. Increased Neuron Number and Head Size in Autism. JAMA 2011, 306, 2031. [Google Scholar] [CrossRef] [PubMed]
- Heitz, U.; Papmeyer, M.; Studerus, E.; Egloff, L.; Ittig, S.; Andreou, C.; Vogel, T.; Borgwardt, S.; Graf, M.; Eckert, A.; et al. Plasma and Serum Brain-Derived Neurotrophic Factor (BDNF) Levels and Their Association with Neurocognition in at-Risk Mental State, First Episode Psychosis and Chronic Schizophrenia Patients. World J. Biol. Psychiatry 2019, 20, 545–554. [Google Scholar] [CrossRef]
- Galvez-Contreras, A.Y.; Campos-Ordonez, T.; Lopez-Virgen, V.; Gomez-Plascencia, J.; Ramos-Zuniga, R.; Gonzalez-Perez, O. Growth Factors as Clinical Biomarkers of Prognosis and Diagnosis in Psychiatric Disorders. Cytokine Growth Factor Rev. 2016, 32, 85–96. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A Glial Cell Line-Derived Neurotrophic Factor for Midbrain Dopaminergic Neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Rocha, S.M.; Cristovão, A.C.; Campos, F.L.; Fonseca, C.P.; Baltazar, G. Astrocyte-Derived GDNF Is a Potent Inhibitor of Microglial Activation. Neurobiol. Dis. 2012, 47, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Paratcha, G.; Ledda, F.; Ibáñez, C.F. The Neural Cell Adhesion Molecule NCAM Is an Alternative Signaling Receptor for GDNF Family Ligands. Cell 2003, 113, 867–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosrat, C.A.; Tomac, A.; Lindqvist, E.; Lindskog, S.; Humpel, C.; Strömberg, I.; Ebendal, T.; Hoffer, B.J.; Olson, L. Cellular Expression of GDNF MRNA Suggests Multiple Functions inside and Outside the Nervous System. Cell Tissue Res. 1996, 286, 191–207. [Google Scholar] [CrossRef]
- Choi-Lundberg, D.L.; Bohn, M.C. Ontogeny and Distribution of Glial Cell Line-Derived Neurotrophic Factor (GDNF) MRNA in Rat. Dev. Brain Res. 1995, 85, 80–88. [Google Scholar] [CrossRef]
- Lei, Z.; Jiang, Y.; Li, T.; Zhu, J.; Zeng, S. Signaling of Glial Cell Line-Derived Neurotrophic Factor and Its Receptor GFRα1 Induce Nurr1 and Pitx3 to Promote Survival of Grafted Midbrain-Derived Neural Stem Cells in a Rat Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2011, 70, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Yurek, D.M.; Flectcher, A.M.; Kowalczyk, T.H.; Padegimas, L.; Cooper, M.J. Compacted DNA Nanoparticle Gene Transfer of GDNF to the Rat Striatum Enhances the Survival of Grafted Fetal Dopamine Neurons. Cell Transpl. 2009, 18, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boger, H.A.; Middaugh, L.D.; Zaman, V.; Hoffer, B.; Granholm, A.-C. Differential Effects of the Dopamine Neurotoxin MPTP in Animals with a Partial Deletion of the GDNF Receptor, GFRα1, Gene. Brain Res. 2008, 1241, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Palasz, E.; Niewiadomski, W.; Gasiorowska, A.; Mietelska-Porowska, A.; Niewiadomska, G. Neuroplasticity and Neuroprotective Effect of Treadmill Training in the Chronic Mouse Model of Parkinson’s Disease. Neural Plast. 2019, 2019, 8215017. [Google Scholar] [CrossRef] [Green Version]
- Hunot, S.; Bernard, V.; Faucheux, B.; Boissière, F.; Leguern, E.; Brana, C.; Gautris, P.P.; Guérin’s, J.; Bloch, B.; Agid, Y.; et al. Glial Cell Line-Derived Neurotrophic Factor (GDNF) Gene Expression in the Human Brain: A Post Mortem in Situ Hybridization Study with Special Reference to Parkinson’s Disease. J. Neural. Transm. 1996, 103, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.B.; Siegel, G.J.; Lee, J.M. Depletion of Glial Cell Line-Derived Neurotrophic Factor in Substantia Nigra Neurons of Parkinson’s Disease Brain. J. Chem. Neuroanat. 2001, 21, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis Control by the Unfolded Protein Response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arancibia, D.; Zamorano, P.; Andrés, M.E. CDNF Induces the Adaptive Unfolded Protein Response and Attenuates Endoplasmic Reticulum Stress-Induced Cell Death. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhao, H.; Zhang, W.; Liu, B.; Liu, Y.; Guo, Y.; Nie, L. Overexpression of Conserved Dopamine Neurotrophic Factor (CDNF) in Astrocytes Alleviates Endoplasmic Reticulum Stress-Induced Cell Damage and Inflammatory Cytokine Secretion. Biochem. Biophys. Res. Commun. 2013, 435, 34–39. [Google Scholar] [CrossRef]
- Voutilainen, M.H.; Bäck, S.; Peränen, J.; Lindholm, P.; Raasmaja, A.; Männistö, P.T.; Saarma, M.; Tuominen, R.K. Chronic Infusion of CDNF Prevents 6-OHDA-Induced Deficits in a Rat Model of Parkinson’s Disease. Exp. Neurol. 2011, 228, 99–108. [Google Scholar] [CrossRef]
- Lindholm, P.; Voutilainen, M.H.; Laurén, J.; Peränen, J.; Leppänen, V.-M.; Andressoo, J.-O.; Lindahl, M.; Janhunen, S.; Kalkkinen, N.; Timmusk, T.; et al. Novel Neurotrophic Factor CDNF Protects and Rescues Midbrain Dopamine Neurons in Vivo. Nature 2007, 448, 73–77. [Google Scholar] [CrossRef]
- Aloe, L. Rita Levi-Montalcini: The Discovery of Nerve Growth Factor and Modern Neurobiology. Trends Cell Biol. 2004, 14, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, G.; Mietelska-Porowska, A.; Mazurkiewicz, M. The Cholinergic System, Nerve Growth Factor and the Cytoskeleton. Behav. Brain Res. 2011, 221, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Dreyfus, C.F. Oligodendrocytes as Providers of Growth Factors. J. Neurosci. Res. 2002, 68, 647–654. [Google Scholar] [CrossRef]
- Niewiadomska, G.; Komorowski, S.; Baksalerska-Pazera, M. Amelioration of Cholinergic Neurons Dysfunction in Aged Rats Depends on the Continuous Supply of NGF. Neurobiol. Aging 2002, 23, 601–613. [Google Scholar] [CrossRef]
- Cuello, A.; Bruno, A.; Bell, K.F. NGF-Cholinergic Dependency in Brain Aging, MCI and Alzheimers Disease. Curr. Alzheimer Res. 2007, 4, 351–358. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Lin, Y.-C.; Sun, C.-K.; Huang, S.-C.; Lee, P.-H.; Kao, Y.-H. Up-Regulation of Nerve Growth Factor in Cholestatic Livers and Its Hepatoprotective Role against Oxidative Stress. PLoS ONE 2014, 9, e112113. [Google Scholar] [CrossRef]
- Minnone, G.; De Benedetti, F.; Bracci-Laudiero, L. NGF and Its Receptors in the Regulation of Inflammatory Response. Int. J. Mol. Sci. 2017, 18, 1028. [Google Scholar] [CrossRef] [Green Version]
- McKelvey, L.; Shorten, G.D.; O’Keeffe, G.W. Nerve Growth Factor-Mediated Regulation of Pain Signalling and Proposed New Intervention Strategies in Clinical Pain Management. J. Neurochem. 2013, 124, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; McMahon, S.B. Neurotrophins: Mediators and Modulators of Pain. Annu. Rev. Neurosci. 2006, 29, 507–538. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Tiveron, C.; Amato, G.; Vignone, D.; Cattaneo, A. Peripheral Neutralization of Nerve Growth Factor Induces Immunosympathectomy and Central Neurodegeneration in Transgenic Mice. J. Alzheimer’s Dis. 2010, 20, 527–546. [Google Scholar] [CrossRef]
- Tiveron, C.; Fasulo, L.; Capsoni, S.; Malerba, F.; Marinelli, S.; Paoletti, F.; Piccinin, S.; Scardigli, R.; Amato, G.; Brandi, R.; et al. ProNGF\NGF Imbalance Triggers Learning and Memory Deficits, Neurodegeneration and Spontaneous Epileptic-like Discharges in Transgenic Mice. Cell Death Differ. 2013, 20, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Brandi, R.; Arisi, I.; D’Onofrio, M.; Cattaneo, A. A Dual Mechanism Linking NGF/ProNGF Imbalance and Early Inflammation to Alzheimer’s Disease Neurodegeneration in the AD11 Anti-NGF Mouse Model. CNS Neurol. Disord. Drug Targets 2011, 10, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Increased ProNGF Levels in Subjects with Mild Cognitive Impairment and Mild Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2004, 63, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Salehi, A.; Verhaagen, J.; Dijkhuizen, P.A.; Swaab, D.F. Co-Localization of High-Affinity Neurotrophin Receptors in Nucleus Basalis of Meynert Neurons and Their Differential Reduction in Alzheimer’s Disease. Neuroscience 1996, 75, 373–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, S.A.; Mufson, E.J.; Weingartner, J.A.; Skau, K.A.; Crutcher, K.A. Nerve Growth Factor in Alzheimer’s Disease: Increased Levels throughout the Brain Coupled with Declines in Nucleus Basalis. J. Neurosci. 1995, 15, 6213–6221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbach, G.J.; Hellweg, R.; Haas, C.A.; Del Turco, D.; Deicke, U.; Abramowski, D.; Jucker, M.; Staufenbiel, M.; Deller, T. Induction of Brain-Derived Neurotrophic Factor in Plaque-Associated Glial Cells of Aged APP23 Transgenic Mice. J. Neurosci. 2004, 24, 2421–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo-Figueroa, M.; Bonilla, S.; Gutiérrez, F.; Pascual, A.; López-Barneo, J. GDNF Is Predominantly Expressed in the PV+ Neostriatal Interneuronal Ensemble in Normal Mouse and after Injury of the Nigrostriatal Pathway. J. Neurosci. 2012, 32, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Schwartz, J.P. Gene Expression Profiles of Reactive Astrocytes in Dopamine-Depleted Striatum. Brain Pathol. 2006, 14, 275–280. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “Quad-partite” Synapse: Microglia-synapse Interactions in the Developing and Mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Casati, M.E.; Murtie, J.C.; Rio, C.; Stankovic, K.; Liberman, M.C.; Corfas, G. Nonneuronal Cells Regulate Synapse Formation in the Vestibular Sensory Epithelium via ErbB-Dependent BDNF Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17005–17010. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, M.-J.; Trudeau, L.-E. GDNF Enhances the Synaptic Efficacy of Dopaminergic Neurons in Culture. Eur. J. Neurosci. 2000, 12, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, I.; Lukomska, B. The Role of Astrocytes in the Physiology and Pathology of the Central Nervous System. Acta Neurobiol. Exp. (Wars) 2006, 66, 343–358. [Google Scholar] [PubMed]
- Jiang, Y.; Wei, N.; Lu, T.; Zhu, J.; Xu, G.; Liu, X. Intranasal Brain-Derived Neurotrophic Factor Protects Brain from Ischemic Insult via Modulating Local Inflammation in Rats. Neuroscience 2011, 172, 398–405. [Google Scholar] [CrossRef]
- Zhao, H.; Cheng, L.; Liu, Y.; Zhang, W.; Maharjan, S.; Cui, Z.; Wang, X.; Tang, D.; Nie, L. Mechanisms of Anti-Inflammatory Property of Conserved Dopamine Neurotrophic Factor: Inhibition of JNK Signaling in Lipopolysaccharide-Induced Microglia. J. Mol. Neurosci. 2014, 52, 186–192. [Google Scholar] [CrossRef]
- Rickert, U.; Grampp, S.; Wilms, H.; Spreu, J.; Knerlich-Lukoschus, F.; Held-Feindt, J.; Lucius, R. Glial Cell Line-Derived Neurotrophic Factor Family Members Reduce Microglial Activation via Inhibiting P38MAPKs-Mediated Inflammatory Responses. J. Neurodegener. Dis. 2014, 2014, 369468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Haney, M.J.; Jin, Y.S.; Uvarov, O.; Vinod, N.; Lee, Y.Z.; Langworthy, B.; Fine, J.P.; Rodriguez, M.; El-Hage, N.; et al. GDNF-Expressing Macrophages Restore Motor Functions at a Severe Late-Stage, and Produce Long-Term Neuroprotective Effects at an Early-Stage of Parkinson’s Disease in Transgenic Parkin Q311X(A) Mice. J. Control. Release 2019, 315, 139–149. [Google Scholar] [CrossRef]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF Steers Microglia toward a Neuroprotective Phenotype. Glia 2018, 66, 1395–1416. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Deng, G.; Huang, H. The Activation of BDNF Reduced Inflammation in a Spinal Cord Injury Model by TrkB/P38 MAPK Signaling. Exp. Ther. Med. 2018, 17, 1688–1696. [Google Scholar] [CrossRef] [Green Version]
- Chou, A.-K.; Yang, M.-C.; Tsai, H.-P.; Chai, C.-Y.; Tai, M.-H.; Kwan, A.-L.; Hong, Y.-R. Adenoviral-Mediated Glial Cell Line–Derived Neurotrophic Factor Gene Transfer Has a Protective Effect on Sciatic Nerve Following Constriction-Induced Spinal Cord Injury. PLoS ONE 2014, 9, e92264. [Google Scholar] [CrossRef]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Hetman, M.; Hsuan, S.-L.; Habas, A.; Higgins, M.J.; Xia, Z. ERK1/2 Antagonizes Glycogen Synthase Kinase-3β-Induced Apoptosis in Cortical Neurons. J. Biol. Chem. 2002, 277, 49577–49584. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jope, R.S. Is Glycogen Synthase Kinase-3 a Central Modulator in Mood Regulation? Neuropsychopharmacology 2010, 35, 2143–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xiang, Y.; Wang, X.; Zhu, L.; Li, H.; Wang, S.; Pan, X.; Zhao, H. Cerebral Dopamine Neurotrophic Factor Protects Microglia by Combining with AKT and by Regulating FoxO1/MTOR Signaling during Neuroinflammation. Biomed. Pharmacother. 2019, 109, 2278–2284. [Google Scholar] [CrossRef]
- Bruna, B.; Lobos, P.; Herrera-Molina, R.; Hidalgo, C.; Paula-Lima, A.; Adasme, T. The Signaling Pathways Underlying BDNF-Induced Nrf2 Hippocampal Nuclear Translocation Involve ROS, RyR-Mediated Ca2+ Signals, ERK and PI3K. Biochem. Biophys. Res. Commun. 2018, 505, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Eremin, D.V.; Ilchibaeva, T.V.; Tsybko, A.S. Cerebral Dopamine Neurotrophic Factor (CDNF): Structure, Functions, and Therapeutic Potential. Biochemistry (Moscow) 2021, 86, 852–866. [Google Scholar] [CrossRef] [PubMed]
- Fodelianaki, G.; Lansing, F.; Bhattarai, P.; Troullinaki, M.; Zeballos, M.A.; Charalampopoulos, I.; Gravanis, A.; Mirtschink, P.; Chavakis, T.; Alexaki, V.I. Nerve Growth Factor Modulates LPS—Induced Microglial Glycolysis and Inflammatory Responses. Exp. Cell Res. 2019, 377, 10–16. [Google Scholar] [CrossRef]
- Albert, K.; Raymundo, D.P.; Panhelainen, A.; Eesmaa, A.; Shvachiy, L.; Araújo, G.R.; Chmielarz, P.; Yan, X.; Singh, A.; Cordeiro, Y.; et al. Cerebral Dopamine Neurotrophic Factor Reduces α-Synuclein Aggregation and Propagation and Alleviates Behavioral Alterations in Vivo. Mol. Ther. 2021, 29, 2821–2840. [Google Scholar] [CrossRef]
- Qing, J.; Liu, X.; Wu, Q.; Zhou, M.; Zhang, Y.; Mazhar, M.; Huang, X.; Wang, L.; He, F. Hippo/YAP Pathway Plays a Critical Role in Effect of GDNF Against Aβ-Induced Inflammation in Microglial Cells. DNA Cell Biol. 2020, 39, 1064–1071. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like Receptor–Mediated Cytokine Production Is Differentially Regulated by Glycogen Synthase Kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Grimes, C.A.; Jope, R.S. CREB DNA Binding Activity Is Inhibited by Glycogen Synthase Kinase-3β and Facilitated by Lithium. J. Neurochem. 2001, 78, 1219–1232. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Pan, B.-S.; Tsai, S.-F.; Chiang, Y.-T.; Huang, B.-M.; Mo, F.-E.; Kuo, Y.-M. BDNF Reverses Aging-Related Microglial Activation. J. Neuroinflammation 2020, 17, 210. [Google Scholar] [CrossRef]
- Shenkar, R.; Yum, H.-K.; Arcaroli, J.; Kupfner, J.; Abraham, E. Interactions between CBP, NF-ΚB, and CREB in the Lungs after Hemorrhage and Endotoxemia. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2001, 281, L418–L426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, G.; Mi, Y.; Wang, Y.; Li, R.; Huang, S.; Li, X.; Liu, X. Neuroprotective Action of Tea Polyphenols on Oxidative Stress-Induced Apoptosis through the Activation of the TrkB/CREB/BDNF Pathway and Keap1/Nrf2 Signaling Pathway in SH-SY5Y Cells and Mice Brain. Food Funct. 2017, 8, 4421–4432. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-ΚB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, Z.; Liu, C.; Zhao, Y.; Chen, Y. Activated Microglia Provide a Neuroprotective Role by Balancing Glial Cell-Line Derived Neurotrophic Factor and Tumor Necrosis Factor-α Secretion after Subacute Cerebral Ischemia. Int. J. Mol. Med. 2013, 31, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Guo, S.; Lu, S.; Zhou, J.; Li, J.; Xia, S. Ultrasound-Induced Release of GDNF from Lipid Coated Microbubbles Injected into Striatum Reduces Hypoxic–Ischemic Injury in Neonatal Rats. Brain Res. Bull. 2012, 88, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Sahu, M.R.; Mondal, A.C. Neuronal Hippo Signaling: From Development to Diseases. Dev. Neurobiol. 2021, 81, 92–109. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Chen, A.; Fa, Z.; Ding, Z.; Xie, J.; Sun, Y.; Zhang, R.; Wang, Q. Adipose-Derived Stem Cells Modulate BV2 Microglial M1/M2 Polarization by Producing GDNF. Stem Cells Dev. 2020, 29, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Tseng, K.-Y.; Wu, J.-S.; Chen, Y.-H.; Airavaara, M.; Cheng, C.-Y.; Ma, K.-H. Modulating Microglia/Macrophage Activation by CDNF Promotes Transplantation of Fetal Ventral Mesencephalic Graft Survival and Function in a Hemiparkinsonian Rat Model. Biomedicines 2022, 10, 1446. [Google Scholar] [CrossRef]
- Lindahl, M.; Chalazonitis, A.; Palm, E.; Pakarinen, E.; Danilova, T.; Pham, T.D.; Setlik, W.; Rao, M.; Võikar, V.; Huotari, J.; et al. Cerebral Dopamine Neurotrophic Factor–Deficiency Leads to Degeneration of Enteric Neurons and Altered Brain Dopamine Neuronal Function in Mice. Neurobiol. Dis. 2020, 134, 104696. [Google Scholar] [CrossRef]
- Freund, V.; Pons, F.; Joly, V.; Mathieu, E.; Martinet, N.; Frossard, N. Upregulation of Nerve Growth Factor Expression by Human Airway Smooth Muscle Cells in Inflammatory Conditions. Eur. Respir. J. 2002, 20, 458–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelz, M.; Mantyh, P.; Malfait, A.-M.; Farrar, J.; Yaksh, T.; Tive, L.; Viktrup, L. Nerve Growth Factor Antibody for the Treatment of Osteoarthritis Pain and Chronic Low-Back Pain. Pain 2019, 160, 2210–2220. [Google Scholar] [CrossRef] [Green Version]
- Niewiadomska, G.; Baksalerska-Pazera, M.; Gasiorowska, A.; Mietelska, A. Nerve Growth Factor Differentially Affects Spatial and Recognition Memory in Aged Rats. Neurochem. Res. 2006, 31, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Weigand, L.A.; Kwong, K.; Myers, A.C. The Effects of Nerve Growth Factor on Nicotinic Synaptic Transmission in Mouse Airway Parasympathetic Neurons. Am. J. Respir. Cell Mol. Biol. 2015, 53, 443–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manca, A.; Capsoni, S.; di Luzio, A.; Vignone, D.; Malerba, F.; Paoletti, F.; Brandi, R.; Arisi, I.; Cattaneo, A.; Levi-Montalcini, R. Nerve Growth Factor Regulates Axial Rotation during Early Stages of Chick Embryo Development. Proc. Natl. Acad. Sci. USA 2012, 109, 2009–2014. [Google Scholar] [CrossRef] [Green Version]
- Tuszynski, M.H. Intraparenchymal NGF Infusions Rescue Degenerating Cholinergic Neurons. Cell Transpl. 2000, 9, 629–636. [Google Scholar] [CrossRef]
- Terrando, N.; Yang, T.; Ryu, J.K.; Newton, P.T.; Monaco, C.; Feldmann, M.; Ma, D.; Akassoglou, K.; Maze, M. Stimulation of the A7 Nicotinic Acetylcholine Receptor Protects against Neuroinflammation after Tibia Fracture and Endotoxemia in Mice. Mol. Med. 2014, 20, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Prencipe, G.; Minnone, G.; Strippoli, R.; de Pasquale, L.; Petrini, S.; Caiello, I.; Manni, L.; de Benedetti, F.; Bracci-Laudiero, L. Nerve Growth Factor Downregulates Inflammatory Response in Human Monocytes through TrkA. J. Immunol. 2014, 192, 3345–3354. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, F.; Siemen, D.; Mawrin, C.; Horn, T.; Dietzmann, K. The Neurotrophin Receptor TrkB Is Colocalized to Mitochondrial Membranes. Int. J. Biochem. Cell Biol. 2006, 38, 610–620. [Google Scholar] [CrossRef]
- Markham, A.; Cameron, I.; Bains, R.; Franklin, P.; Kiss, J.P.; Schwendimann, L.; Gressens, P.; Spedding, M. Brain-Derived Neurotrophic Factor-Mediated Effects on Mitochondrial Respiratory Coupling and Neuroprotection Share the Same Molecular Signalling Pathways. Eur. J. Neurosci. 2012, 35, 366–374. [Google Scholar] [CrossRef]
- Armijo-Weingart, L.; Ketschek, A.; Sainath, R.; Pacheco, A.; Smith, G.M.; Gallo, G. Neurotrophins Induce Fission of Mitochondria along Embryonic Sensory Axons. Elife 2019, 8, e49494. [Google Scholar] [CrossRef] [PubMed]
- Chada, S.R.; Hollenbeck, P.J. Nerve Growth Factor Signaling Regulates Motility and Docking of Axonal Mitochondria. Curr. Biol. 2004, 14, 1272–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial Inner Membrane Permeabilisation Enables MtDNA Release during Apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Fritsch, L.E.; Kelly, C.; Pickrell, A.M. The Role of STING Signaling in Central Nervous System Infection and Neuroinflammatory Disease. WIREs Mech. Dis. 2023, e1597. [Google Scholar] [CrossRef]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 Recruitment to STING Activates Both IRF3 and NF-ΚB That Mediate Immune Defense against Tumors and Viral Infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced MtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhu, Y.; Li, Y.; Ding, X.; Ma, W.; Han, X.; Wang, B. BDNF-Mediated Mitophagy Alleviates High-Glucose-Induced Brain Microvascular Endothelial Cell Injury. Apoptosis 2019, 24, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Ji, Y.-S.; Sun, X.; Liu, X.-H.; Chen, Z.-Y. Brain-Derived Neurotrophic Factor (BDNF)-Induced Mitochondrial Motility Arrest and Presynaptic Docking Contribute to BDNF-Enhanced Synaptic Transmission. J. Biol. Chem. 2014, 289, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- McManus, M.J.; Franklin, J.L. Dissociation of JNK Activation from Elevated Levels of Reactive Oxygen Species, Cytochrome c Release, and Cell Death in NGF-Deprived Sympathetic Neurons. Mol. Neurobiol. 2018, 55, 382–389. [Google Scholar] [CrossRef]
- Kirkland, R.A.; Saavedra, G.M.; Franklin, J.L. Rapid Activation of Antioxidant Defenses by Nerve Growth Factor Suppresses Reactive Oxygen Species during Neuronal Apoptosis: Evidence for a Role in Cytochrome c Redistribution. J. Neurosci. 2007, 27, 11315–11326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Hu, W.; Yin, S.; Lu, X.; Zuo, W.; Ge, S.; Xu, Y. NGF Protects against Oxygen and Glucose Deprivation-Induced Oxidative Stress and Apoptosis by up-Regulation of HO-1 through MEK/ERK Pathway. Neurosci. Lett. 2017, 641, 8–14. [Google Scholar] [CrossRef]
- Molinari, C.; Morsanuto, V.; Ruga, S.; Notte, F.; Farghali, M.; Galla, R.; Uberti, F. The Role of BDNF on Aging-Modulation Markers. Brain Sci. 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of Brain-Derived Neurotrophic Factor across the Blood–Brain Barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier Drug Targeting Enables Neuroprotection in Brain Ischemia Following Delayed Intravenous Administration of Neurotrophins. Adv. Exp. Med. Biol. 2002, 513, 397–430. [Google Scholar] [PubMed]
- Saragovi, H.U.; Gehring, K. Development of Pharmacological Agents for Targeting Neurotrophins and Their Receptors. Trends Pharmacol. Sci. 2000, 21, 93–98. [Google Scholar] [CrossRef]
- Miller, R.G.; Bryan, W.W.; Dietz, M.A.; Munsat, T.L.; Petajan, J.H.; Smith, S.A.; Goodpasture, J.C. Toxicity and Tolerability of Recombinant Human Ciliary Neurotrophic Factor in Patients with Amyotrophic Lateral Sclerosis. Neurology 1996, 47, 1329–1331. [Google Scholar] [CrossRef]
- Miller, R.G.; Petajan, J.H.; Bryan, W.W.; Armon, C.; Barohn, R.J.; Goodpasture, J.C.; Hoagland, R.J.; Parry, G.J.; Ross, M.A.; Stromatt, S.C. A Placebo-Controlled Trial of Recombinant Human Ciliary Neurotrophic (RhCNTF) Factor in Amyotrophic Lateral Sclerosis. Ann. Neurol. 1996, 39, 256–260. [Google Scholar] [CrossRef]
- A Double-Blind Placebo-Controlled Clinical Trial of Subcutaneous Recombinant Human Ciliary Neurotrophic Factor (RHCNTF) in Amyotrophic Lateral Sclerosis. Neurology 1996, 46, 1244. [CrossRef]
- Bongioanni, P.; Reali, C.; Sogos, V. Ciliary Neurotrophic Factor (CNTF) for Amyotrophic Lateral Sclerosis or Motor Neuron Disease. Cochrane Database Syst. Rev. 2004, 2004, CD004302. [Google Scholar] [CrossRef]
- A Phase I Study of Recombinant Human Ciliary Neurotrophic Factor (RHCNTF) in Patients with Amyotrophic Lateral Sclerosis. Clin. Neuropharmacol. 1995, 18, 515–532. [CrossRef] [PubMed]
- Nutt, J.G.; Burchiel, K.J.; Comella, C.L.; Jankovic, J.; Lang, A.E.; Laws, E.R.; Lozano, A.M.; Penn, R.D.; Simpson, R.K.; Stacy, M.; et al. Randomized, Double-Blind Trial of Glial Cell Line-Derived Neurotrophic Factor (GDNF) in PD. Neurology 2003, 60, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.K.; Bunnage, M.; Plaha, P.; Svendsen, C.N.; Heywood, P.; Gill, S.S. Intraputamenal Infusion of Glial Cell Line-Derived Neurotrophic Factor in PD: A Two-Year Outcome Study. Ann. Neurol. 2005, 57, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized Controlled Trial of Intraputamenal Glial Cell Line-Derived Neurotrophic Factor Infusion in Parkinson Disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Bachoud-Lévi, A.-C.; Déglon, N.; Nguyen, J.-P.; Bloch, J.; Bourdet, C.; Winkel, L.; Rémy, P.; Goddard, M.; Lefaucheur, J.-P.; Brugières, P.; et al. Neuroprotective Gene Therapy for Huntington’s Disease Using a Polymer Encapsulated BHK Cell Line Engineered to Secrete Human CNTF. Hum. Gene Ther. 2000, 11, 1723–1729. [Google Scholar] [CrossRef]
- Aebischer, P.; Schluep, M.; Déglon, N.; Joseph, J.-M.; Hirt, L.; Heyd, B.; Goddard, M.; Hammang, J.P.; Zurn, A.D.; Kato, A.C.; et al. Intrathecal Delivery of CNTF Using Encapsulated Genetically Modifiedxenogeneic Cells in Amyotrophic Lateral Sclerosis Patients. Nat. Med. 1996, 2, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; U, H.S.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A Phase 1 Clinical Trial of Nerve Growth Factor Gene Therapy for Alzheimer Disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Yang, J.H.; Barba, D.; U, H.-S.; Bakay, R.A.E.; Pay, M.M.; Masliah, E.; Conner, J.M.; Kobalka, P.; Roy, S.; et al. Nerve Growth Factor Gene Therapy. JAMA Neurol. 2015, 72, 1139. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://cordis.europa.eu/project/id/732386 (accessed on 24 February 2023).




{kind=link}
{kind=link}
{kind=link}
| NTF | Physiological Function | Disease | Model | Structure | Cell Type/Expression | References |
|---|---|---|---|---|---|---|
| Brain-derived neurotrophic factor (BDNF) | Binds to tropomyosin receptor kinase B (TrkB); activates phospholipase C-γ (PLCγ) and Ras; induces dendritic growth, development of synapses, survival of neurons; regulates synaptic plasticity | Parkinson’s disease (PD) | PD patients; post-mortem | Substantia nigra pars compacta (SNpc) | Dopaminergic neurons ↓ | [49] |
| Alzheimer’s disease (AD) | APP23 transgenic mice | Cortex | Astrocytes, microglia ↑ | [87] | ||
| Autism spectrum disorder (ASD) | Newborns | Peripheral blood | Dried blood spot samples ↓ | [50] | ||
| Children | Peripheral blood | ↑ | [51] | |||
| Glial cell-derived neurotrophic factor (GDNF) | Binds to GDNF family receptor (GFR)α1, GFRα2, and GFRα3; GDNF/ receptor tyrosine kinase rearranged during transfection (RET) activates mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), and proto-oncogene tyrosine-protein kinase (Src) pathways; GDNF/ neural cell adhesion molecule (NCAM) activates Fyn and FAK protein kinases, stimulates proliferation, cell migration, neurite elongation and neurite branching, Schwann cell migration, and axonal growth | PD | PD patients; post-mortem | SNpc | Neurons | [66] |
| Mesencephalon/striatum | 0 | [65] | ||||
| 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mice model | Striatum | Parvalbumin-positive (PV+) interneurons ↔ | [88] | |||
| Nerve growth factor (NGF) | Binds to tropomyosin receptor kinase A (TrkA); activates MAPK, PLC-γ, and PI3/Akt kinase; stimulates neuronal survival, growth, and differentiation; binds to p75NTR; stimulates c-Jun N-terminal kinase (JNK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ĸB); activates neuronal survival and apoptosis | AD | AD patients; post-mortem | The hippocampus, superior temporal gyrus, superior frontal gyrus, inferior parietal lobule, frontal and occipital cortical poles, cerebellum, amygdala, and putamen | ↑ | [86] |
| Nucleus basalis of Meynert | ↓ | |||||
| Mild cognitive impairment (MCI) and AD patients; post-mortem | Parietal cortex | ↑ | [85] | |||
| PD | 6-hydroxydopamine (6-OHDA) rat model | Striatum | Astrocytes ↑ | [89] | ||
| ASD | Children | Peripheral blood | ↑ | [51] |
| Signaling Pathway | NTFs | Effect | References |
|---|---|---|---|
| Suppression of p38 | Brain-derived neurotrophic factor (BDNF); Glial cell-derived neurotrophic factor (GDNF) | Reduction of inflammation (inhibition of production of inflammatory cytokines and enzymes) | [41,100,101] |
| Stimulation of phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), and extracellular signal-regulated kinase (ERK) | Nerve growth factor (NGF); BDNF; GDNF | Reduction of inflammation, upregulation of antioxidant defense, enhancement of neuronal survival (inhibition of glycogen synthase kinase 3 (GSK3) activity, suppression of nuclear factor kappa-light-chain-enhancer of activated B cells (NFĸB)-dependent transcription of pro-inflammatory cytokine genes, induction of nuclear factor erythroid 2-related factor 2 (Nrf2), increased cAMP response element-binding protein (CREB) activity) | [102,103,104,105,106] |
| Inhibition of c-Jun N-terminal kinase (JNK) | Cerebral dopamine neurotrophic factor (CDNF); BDNF | Regulation of microglia activation | [106,107,108] |
| Unknown | CDNF | Inhibition of α-synuclein aggregation | [109] |
| Stimulation of Hippo/yes-associated protein (YAP) | GDNF | Reduction of amyloid beta (Aβ)-induced inflammation | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palasz, E.; Wilkaniec, A.; Stanaszek, L.; Andrzejewska, A.; Adamczyk, A. Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders. Int. J. Mol. Sci. 2023, 24, 6321. https://doi.org/10.3390/ijms24076321
Palasz E, Wilkaniec A, Stanaszek L, Andrzejewska A, Adamczyk A. Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders. International Journal of Molecular Sciences. 2023; 24(7):6321. https://doi.org/10.3390/ijms24076321
Chicago/Turabian StylePalasz, Ewelina, Anna Wilkaniec, Luiza Stanaszek, Anna Andrzejewska, and Agata Adamczyk. 2023. "Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders" International Journal of Molecular Sciences 24, no. 7: 6321. https://doi.org/10.3390/ijms24076321