
DNA Damage-Mediated Neurotoxicity in Parkinson’s Disease

Abstract
:1. Introduction
2. Evidence for DNA Damage-Mediated Neurodegeneration in PD
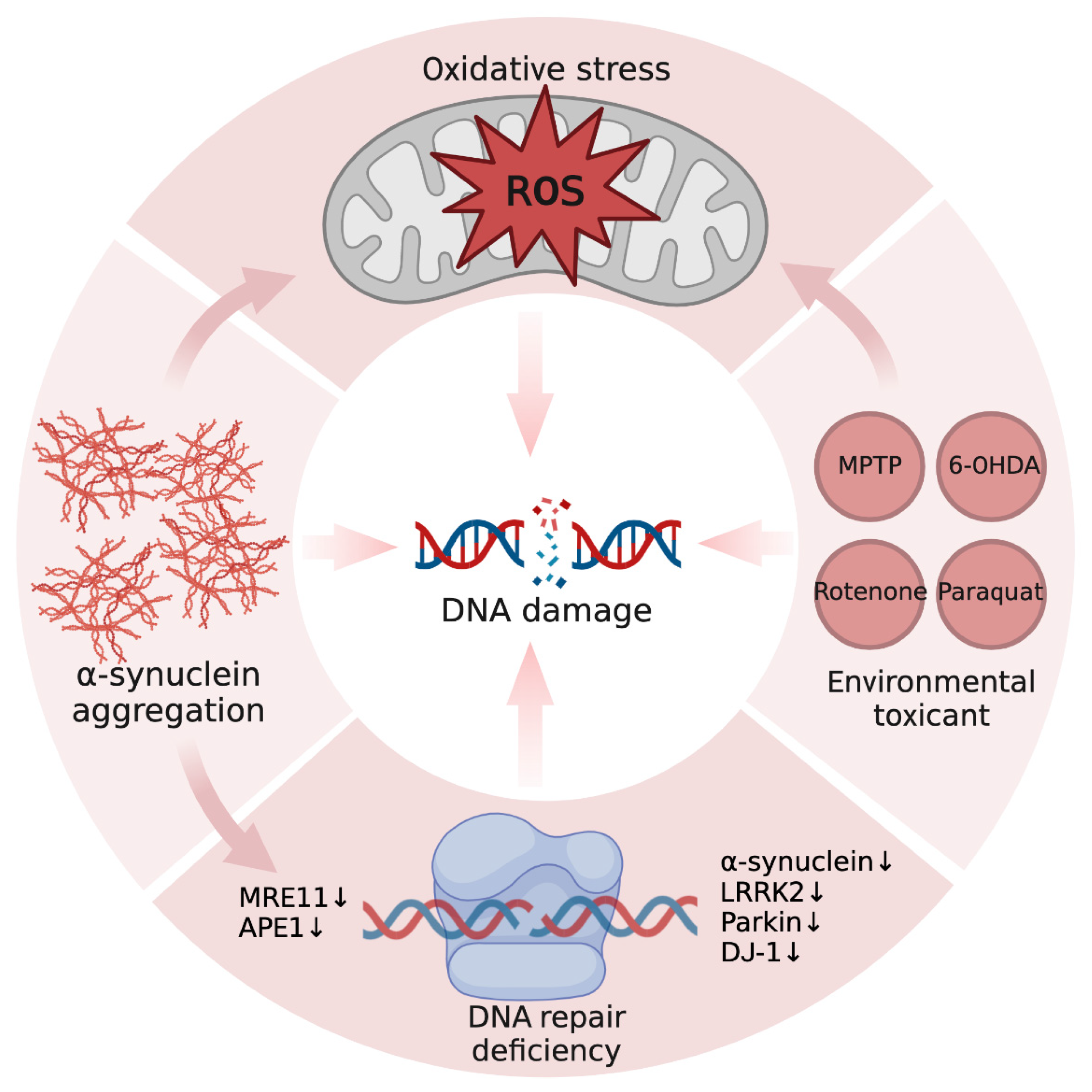
3. Cause of DNA Damage in PD
3.1. Oxidative Stress
3.2. Protein Aggregation
3.3. DNA Repair Deficiency
3.4. Environmental Toxicant
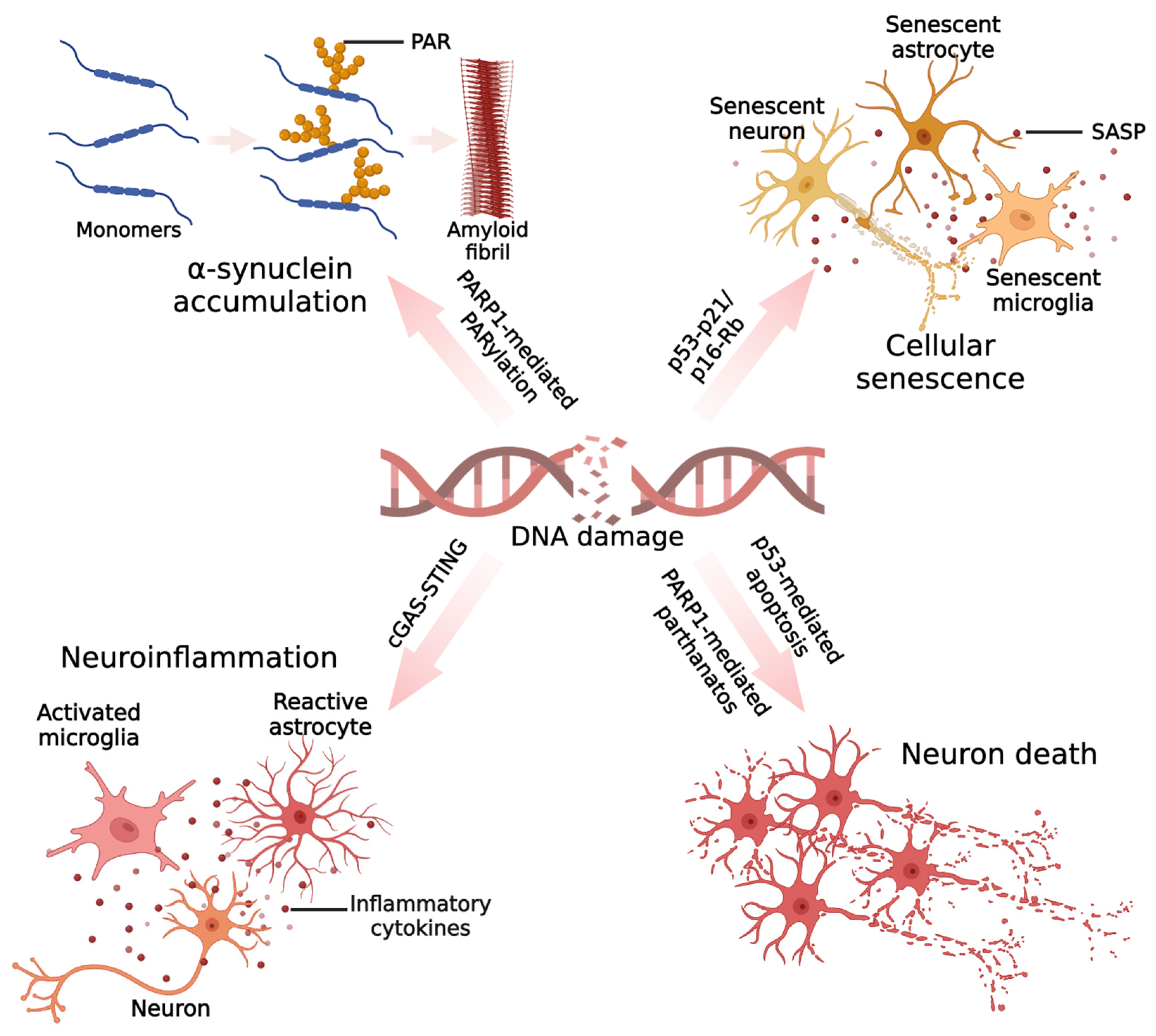
4. Mechanisms of DNA Damage-Mediated Neurotoxicity in PD
4.1. Protein Aggregation
4.2. Cellular Senescence
4.3. Neuroinflammation
4.3.1. Microglia
4.3.2. Astrocytes
4.3.3. Neurons
4.4. Cell Death
4.4.1. Apoptosis
4.4.2. Parthanatos
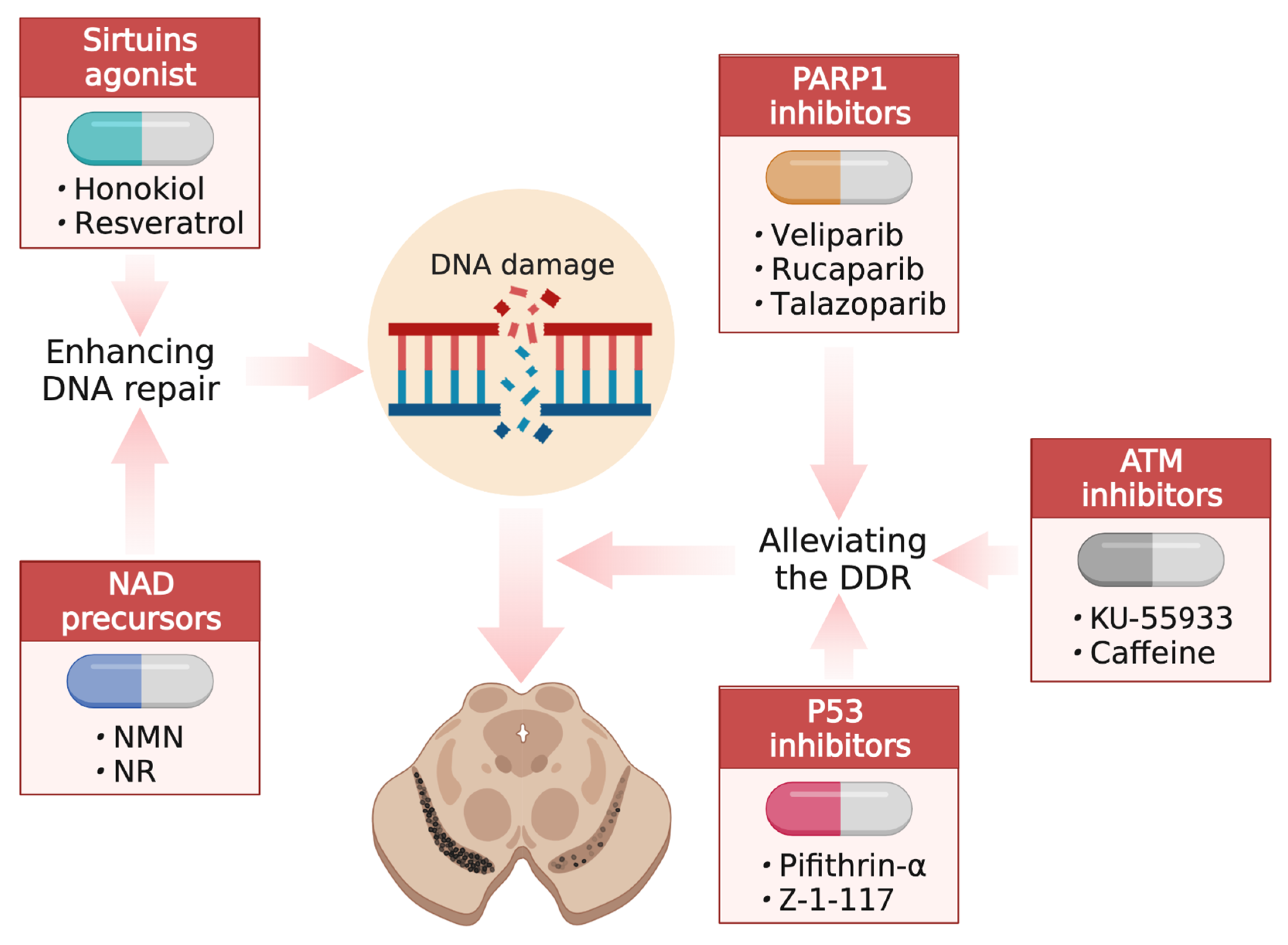
5. Developing Interventions through DNA Damage and Repair
5.1. Enhancing DNA Repair
5.2. Alleviating the DDR
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 559–572. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef] [Green Version]
- Welch, G.; Tsai, L.H. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022, 23, e54217. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Zhao, K.; Tang, D. Contributions of DNA Damage to Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lautrup, S.; Caponio, D.; Zhang, J.; Fang, E.F. DNA Damage-Induced Neurodegeneration in Accelerated Ageing and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6748. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Wang, Z.X.; Ying, C.Z.; Zhang, B.R.; Pu, J.L. Decoding the Role of Familial Parkinson’s Disease-Related Genes in DNA Damage and Repair. Aging Dis. 2022, 13, 1405–1412. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Sanders, L.H. DNA damage and repair in Parkinson’s disease: Recent advances and new opportunities. J. Neurosci. Res. 2021, 99, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.; Atkin, J.D. DNA Damage, Defective DNA Repair, and Neurodegeneration in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2022, 14, 786420. [Google Scholar] [CrossRef]
- Sun, Y.; Curle, A.J.; Haider, A.M.; Balmus, G. The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 2020, 64, 847–861. [Google Scholar] [CrossRef]
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018, 9, 818. [Google Scholar] [CrossRef] [Green Version]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yu, T.; Liu, Y.; Yan, J.; Guo, Y.; Jing, Y.; Yang, X.; Song, Y.; Tian, Y. DNA damage preceding dopamine neuron degeneration in A53T human α-synuclein transgenic mice. Biochem. Biophys. Res. Commun. 2016, 481, 104–110. [Google Scholar] [CrossRef]
- Eilam, R.; Peter, Y.; Elson, A.; Rotman, G.; Shiloh, Y.; Groner, Y.; Segal, M. Selective loss of dopaminergic nigro-striatal neurons in brains of Atm-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 12653–12656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirshner, M.; Galron, R.; Frenkel, D.; Mandelbaum, G.; Shiloh, Y.; Wang, Z.-Q.; Barzilai, A. Malfunctioning DNA damage response (DDR) leads to the degeneration of nigro-striatal pathway in mouse brain. J. Mol. Neurosci. 2012, 46, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; van Ijcken, W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S.; et al. Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson’s Disease. Cell Rep. 2016, 15, 1866–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem. Int. 2012, 61, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef] [Green Version]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef]
- Kikuchia, A.; Takeda, A.; Onoderaa, H.; Kimparaab, T.; Hisanagac, K.; Satoc, N.; Nunomurad, A.; Castellani, R.J.; Perry, G.; Smith, M.A.; et al. Systemic increase of oxidative nucleic acid damage in Parkinson’s disease and multiple system atrophy. Neurobiol. Dis. 2002, 9, 244–248. [Google Scholar] [CrossRef] [Green Version]
- El-Saadi, M.W.; Tian, X.; Grames, M.; Ren, M.; Keys, K.; Li, H.; Knott, E.; Yin, H.; Huang, S.; Lu, X.-H. Tracing brain genotoxic stress in Parkinson’s disease with a novel single-cell genetic sensor. Sci. Adv. 2022, 8, eabd1700. [Google Scholar] [CrossRef]
- Bernstein, A.I.; Garrison, S.P.; Zambetti, G.P.; O’Malley, K.L. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol. Neurodegener. 2011, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Oikawa, S.; Umemura, S.; Hirosawa, I.; Kawanishi, S. Mechanism of metal-mediated DNA damage and apoptosis induced by 6-hydroxydopamine in neuroblastoma SH-SY5Y cells. Free. Radic. Res. 2008, 42, 651–660. [Google Scholar] [CrossRef]
- Chen, Q.; Niu, Y.; Zhang, R.; Guo, H.; Gao, Y.; Li, Y.; Liu, R. The toxic influence of paraquat on hippocampus of mice: Involvement of oxidative stress. Neurotoxicology 2010, 31, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Xiao, J.; Hori, R.; Alway, S.E.; Khan, M.M. Brain Selective Estrogen Treatment Protects Dopaminergic Neurons and Preserves Behavioral Function in MPTP-induced Mouse Model of Parkinson’s Disease. J. Neuroimmune. Pharmacol. 2021, 16, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Rekaik, H.; de Thé, F.-X.B.; Fuchs, J.; Massiani-Beaudoin, O.; Prochiantz, A.; Joshi, R.L. Engrailed Homeoprotein Protects Mesencephalic Dopaminergic Neurons from Oxidative Stress. Cell Rep. 2015, 13, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Alvira, D.; Yeste-Velasco, M.; Folch, J.; Casadesús, G.; Smith, M.A.; Pallàs, M.; Camins, A. Neuroprotective effects of caffeine against complex I inhibition-induced apoptosis are mediated by inhibition of the Atm/p53/E2F-1 path in cerebellar granule neurons. J. Neurosci. Res. 2007, 85, 3079–3088. [Google Scholar] [CrossRef]
- Li, D.W.; Li, G.R.; Zhang, B.L.; Feng, J.J.; Zhao, H. Damage to dopaminergic neurons is mediated by proliferating cell nuclear antigen through the p53 pathway under conditions of oxidative stress in a cell model of Parkinson’s disease. Int. J. Mol. Med. 2016, 37, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camins, A.; Pizarro, J.G.; Alvira, D.; Gutierrez-Cuesta, J.; De La Torre, A.V.; Folch, J.; Sureda, F.X.; Verdaguer, E.; Junyent, F.; Jordán, J.; et al. Activation of ataxia telangiectasia muted under experimental models and human Parkinson’s disease. Cell. Mol. Life Sci. 2010, 67, 3865–3882. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Grammatopoulos, T.N.; Altmann, S.; Amore, A.; Standaert, D.G.; Hyman, B.T.; Kazantsev, A.G. Pharmacological inhibition of PARP-1 reduces alpha-synuclein- and MPP+-induced cytotoxicity in Parkinson’s disease in vitro models. Biochem. Biophys. Res. Commun. 2007, 357, 596–602. [Google Scholar] [CrossRef]
- Lengyel-Zhand, Z.; Puentes, L.N.; Mach, R.H. PARkinson’s: From cellular mechanisms to potential therapeutics. Pharmacol. Ther. 2022, 230, 107968. [Google Scholar] [CrossRef]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Chen, X.; Xie, C.; Tian, W.; Sun, L.; Zheng, W.; Hawes, S.; Chang, L.; Kung, J.; Ding, J.; Chen, S.; et al. Parkinson’s disease-related Leucine-rich repeat kinase 2 modulates nuclear morphology and genomic stability in striatal projection neurons during aging. Mol. Neurodegener. 2020, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cao, Z.; Zhang, W.; Gu, M.; Zhou, Z.D.; Li, B.; Li, J.; Tan, E.K.; Zeng, L. LRRK2 interacts with ATM and regulates Mdm2-p53 cell proliferation axis in response to genotoxic stress. Hum. Mol. Genet 2017, 26, 4494–4505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, H.M.; Mohamed, A.F.; El-Khatib, A.S.; Khattab, M.M. Kinases control of regulated cell death revealing druggable targets for Parkinson’s disease. Ageing Res. Rev. 2023, 85, 101841. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, S.Y. Regulation of DNA repair by parkin. Biochem. Biophys. Res. Commun. 2009, 382, 321–325. [Google Scholar] [CrossRef]
- Eilam, R.; Peter, Y.; Groner, Y.; Segal, M. Late degeneration of nigro-striatal neurons in ATM−/− mice. Neuroscience 2003, 121, 83–98. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 796–814. [Google Scholar] [CrossRef]
- Vlahopoulos, S.; Adamaki, M.; Khoury, N.; Zoumpourlis, V.; Boldogh, I. Roles of DNA repair enzyme OGG1 in innate immunity and its significance for lung cancer. Pharmacol. Ther. 2019, 194, 59–72. [Google Scholar] [CrossRef]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair. 2011, 10, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Miner, K.M.; Jamenis, A.S.; Bhatia, T.N.; Clark, R.N.; Rajasundaram, D.; Sauvaigo, S.; Mason, D.M.; Posimo, J.M.; Abraham, N.; DeMarco, B.A.; et al. α-synucleinopathy exerts sex-dimorphic effects on the multipurpose DNA repair/redox protein APE1 in mice and humans. Prog. Neurobiol. 2022, 216, 102307. [Google Scholar] [CrossRef]
- Abbotts, R.; Madhusudan, S. Human AP endonuclease 1 (APE1): From mechanistic insights to druggable target in cancer. Cancer Treat. Rev. 2010, 36, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Renaudin, X. Reactive oxygen species and DNA damage response in cancer. Int. Rev. Cell Mol. Biol. 2021, 364, 139–161. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef]
- Wu, L.L.; Chiou, C.C.; Chang, P.Y.; Wu, J.T. Urinary 8-OHdG: A marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin. Chim. Acta 2004, 339, 1–9. [Google Scholar] [CrossRef]
- Campos, P.B.; Paulsen, B.S.; Rehen, S.K. Accelerating neuronal aging in in vitro model brain disorders: A focus on reactive oxygen species. Front. Aging Neurosci. 2014, 6, 292. [Google Scholar] [CrossRef] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [Green Version]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainslie, A.; Huiting, W.; Barazzuol, L.; Bergink, S. Genome instability and loss of protein homeostasis: Converging paths to neurodegeneration? Open. Biol. 2021, 11, 200296. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Tramutola, A.; Cascella, R. Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress. Oxid. Med. Cell Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, T.T. DNA damage and regulation of protein homeostasis. DNA Repair. 2021, 105, 103155. [Google Scholar] [CrossRef]
- Basu, S.; Je, G.; Kim, Y.S. Transcriptional mutagenesis by 8-oxodG in α-synuclein aggregation and the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2015, 47, e179. [Google Scholar] [CrossRef] [Green Version]
- Vasquez, V.; Mitra, J.; Hegde, P.M.; Pandey, A.; Sengupta, S.; Mitra, S.; Rao, K.; Hegde, M.L. Chromatin-Bound Oxidized α-Synuclein Causes Strand Breaks in Neuronal Genomes in in vitro Models of Parkinson’s Disease. J. Alzheimers Dis. 2017, 60, S133–S150. [Google Scholar] [CrossRef]
- Pan, Y.; Zong, Q.; Li, G.; Wu, Z.; Du, T.; Huang, Z.; Zhang, Y.; Ma, K. Nuclear localization of alpha-synuclein affects the cognitive and motor behavior of mice by inducing DNA damage and abnormal cell cycle of hippocampal neurons. Front. Mol. Neurosci. 2022, 15, 1015881. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; You, J.S.; Kim, T.-K.; Ahn, W.J.; Kim, M.J.; Son, K.H.; Ricarte, D.; Ortiz, D.; Lee, S.-J.; Lee, H.-J. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp. Mol. Med. 2022, 54, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Williams, J.S.; Tainer, J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 2007, 85, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Reginato, G.; Cejka, P. The MRE11 complex: A versatile toolkit for the repair of broken DNA. DNA Repair. 2020, 91–92, 102869. [Google Scholar] [CrossRef]
- Thakur, S.; Dhiman, M.; Tell, G.; Mantha, A.K. A review on protein-protein interaction network of APE1/Ref-1 and its associated biological functions. Cell Biochem. Funct. 2015, 33, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Geng, A.; Xue, R. Activated or Impaired: An Overview of DNA Repair in Neurodegenerative Diseases. Aging Dis. 2022, 13, 987–1004. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef]
- Chen, L.; Hou, J.; Zeng, X.; Guo, Q.; Deng, M.; A Kloeber, J.; Tu, X.; Zhao, F.; Wu, Z.; Huang, J.; et al. LRRK2 inhibition potentiates PARP inhibitor cytotoxicity through inhibiting homologous recombination-mediated DNA double strand break repair. Clin. Transl. Med. 2021, 11, e341. [Google Scholar] [CrossRef]
- Kao, S.Y. DNA damage induces nuclear translocation of parkin. J. Biomed. Sci. 2009, 16, 67. [Google Scholar] [CrossRef] [Green Version]
- Kosmider, B.; Lin, C.-R.; Vlasenko, L.; Marchetti, N.; Bolla, S.; Criner, G.J.; Messier, E.; Reisdorph, N.; Powell, R.L.; Madesh, M.; et al. Impaired non-homologous end joining in human primary alveolar type II cells in emphysema. Sci. Rep. 2019, 9, 920. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.; Choi, D.-K.; Nagai, M.; Wu, D.-C.; Nagata, T.; Prou, D.; Wilson, G.L.; Vila, M.; Jackson-Lewis, V.; Dawson, V.L.; et al. Neuronal NOS and cyclooxygenase-2 contribute to DNA damage in a mouse model of Parkinson disease. Free Radic. Biol. Med. 2009, 47, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.H.; Timothy Greenamyre, J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Zhang, Q.; Wang, S.T.; Gu, L.; Yang, H.M.; Liu, L.; Bakshi, R.; Zhang, H. Blockade of metabotropic glutamate receptor 5 protects against DNA damage in a rotenone-induced Parkinson’s disease model. Free Radic. Biol. Med. 2015, 89, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2013, 32, 3789–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Fielder, E.; von Zglinicki, T.; Jurk, D. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J. Alzheimers Dis. 2017, 60, S107–S131. [Google Scholar] [CrossRef]
- Espay, A.J.; Vizcarra, J.A.; Marsili, L.; Lang, A.E.; Simon, D.K.; Merola, A.; Josephs, K.A.; Fasano, A.; Morgante, F.; Savica, R.; et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019, 92, 329–337. [Google Scholar] [CrossRef]
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Prog. Neurobiol. 2017, 155, 171–193. [Google Scholar] [CrossRef]
- Lee, J.H.; Ryu, S.W.; Ender, N.A.; Paull, T.T. Poly-ADP-ribosylation drives loss of protein homeostasis in ATM and Mre11 deficiency. Mol. Cell 2021, 81, 1515–1533.e1515. [Google Scholar] [CrossRef]
- Lee, J.-H.; Mand, M.R.; Kao, C.-H.; Zhou, Y.; Ryu, S.W.; Richards, A.L.; Coon, J.J.; Paull, T.T. ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci. Signal. 2018, 11, eaan5598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huiting, W.; Dekker, S.L.; van der Lienden, J.C.; Mergener, R.; Musskopf, M.K.; Furtado, G.V.; Gerrits, E.; Coit, D.; Oghbaie, M.; Di Stefano, L.H.; et al. Targeting DNA topoisomerases or checkpoint kinases results in an overload of chaperone systems, triggering aggregation of a metastable subproteome. Elife 2022, 11, e70726. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Rifai, O.M.; Bonini, N.M. Poly(ADP-Ribosylation) in Age-Related Neurological Disease. Trends Genet. 2019, 35, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L. Poly(ADP-ribose): A Dynamic Trigger for Biomolecular Condensate Formation. Trends Cell Biol. 2020, 30, 370–383. [Google Scholar] [CrossRef]
- Gao, J.; Mewborne, Q.T.; Girdhar, A.; Sheth, U.; Coyne, A.N.; Punathil, R.; Kang, B.G.; Dasovich, M.; Veire, A.; Hernandez, M.D.; et al. Poly(ADP-ribose) promotes toxicity of C9ORF72 arginine-rich dipeptide repeat proteins. Sci. Transl. Med. 2022, 14, eabq3215. [Google Scholar] [CrossRef] [PubMed]
- Rhine, K.; Dasovich, M.; Yoniles, J.; Badiee, M.; Skanchy, S.; Ganser, L.R.; Ge, Y.; Fare, C.M.; Shorter, J.; Leung, A.K.; et al. Poly(ADP-ribose) drives condensation of FUS via a transient interaction. Mol. Cell 2022, 82, 969–985.e911. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Du, A.; Gu, J.; Duan, G.; Wang, C.; Gui, X.; Ma, Z.; Qian, B.; Deng, X.; Zhang, K.; et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 2019, 29, 233–247. [Google Scholar] [CrossRef] [Green Version]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell 2018, 71, 703–717.e709. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Gracia, P.; Polanco, D.; Tarancón-Díez, J.; Serra, I.; Bracci, M.; Oroz, J.; Laurents, D.V.; García, I.; Cremades, N. Molecular mechanism for the synchronized electrostatic coacervation and co-aggregation of alpha-synuclein and tau. Nat. Commun. 2022, 13, 4586. [Google Scholar] [CrossRef] [PubMed]
- Lipiński, W.P.; Visser, B.S.; Robu, I.; Fakhree, M.A.A.; Lindhoud, S.; Claessens, M.M.A.E.; Spruijt, E. Biomolecular condensates can both accelerate and suppress aggregation of α-synuclein. Sci. Adv. 2022, 8, eabq6495. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.-B.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Fang, Y. New insights of poly(ADP-ribosylation) in neurodegenerative diseases: A focus on protein phase separation and pathologic aggregation. Biochem. Pharmacol. 2019, 167, 58–63. [Google Scholar] [CrossRef]
- Pessina, F.; Gioia, U.; Brandi, O.; Farina, S.; Ceccon, M.; Francia, S.; di Fagagna, F.D. DNA Damage Triggers a New Phase in Neurodegeneration. Trends Genet. 2021, 37, 337–354. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; Croix, C.M.S.; Usas, A.; Vo, N.; et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 Induces p21-Dependent Cellular Senescence in Post-mitotic Dopaminergic Neurons. Cell Stem. Cell 2019, 25, 514–530.e518. [Google Scholar] [CrossRef] [Green Version]
- Martin-Ruiz, C.; Williams-Gray, C.H.; Yarnall, A.J.; Boucher, J.J.; Lawson, R.A.; Wijeyekoon, R.S.; Barker, R.A.; Kolenda, C.; Parker, C.; Burn, D.J.; et al. Senescence and Inflammatory Markers for Predicting Clinical Progression in Parkinson’s Disease: The ICICLE-PD Study. J. Parkinsons. Dis. 2020, 10, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Verma, D.; Seo, B.; Ghosh, A.; Ma, S.-X.; Hernandez-Quijada, K.; Andersen, J.; Ko, H.; Kim, Y.-H. Alpha-Synuclein Preformed Fibrils Induce Cellular Senescence in Parkinson’s Disease Models. Cells 2021, 10, 1694. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.; Song, J.; Yang, Y.; Kim, S.; Kim, J.Y.; Seok, M.; Hwang, I.; Yu, J.; Karmacharya, J.; Maeng, H.; et al. SGK1 inhibition in glia ameliorates pathologies and symptoms in Parkinson disease animal models. EMBO Mol. Med. 2021, 13, e13076. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, R.J.; Avramopoulos, D.; Jantzie, L.L.; McCallion, A.S. Neuroinflammation represents a common theme amongst genetic and environmental risk factors for Alzheimer and Parkinson diseases. J. Neuroinflammation 2022, 19, 223. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Simon, M.; Seluanov, A.; Gorbunova, V. DNA damage and repair in age-related inflammation. Nat. Rev. Immunol. 2023, 23, 75–89. [Google Scholar] [CrossRef]
- Pezone, A.; Olivieri, F.; Napoli, M.V.; Procopio, A.; Avvedimento, E.V.; Gabrielli, A. Inflammation and DNA damage: Cause, effect or both. Nat. Rev. Rheumatol. 2023, 19, 200–211. [Google Scholar] [CrossRef]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Song, X.; AW, T.M.; Ma, F.; Leung, D.; Herrup, K. DNA damage induced neuroinflammation in neurodegenerative disease: Developing topics. Alzheimer’s Dement. 2020, 16, e047445. [Google Scholar] [CrossRef]
- Bourseguin, J.; Cheng, W.; Talbot, E.; Hardy, L.; Lai, J.; Jeffries, A.M.; A Lodato, M.; Lee, E.A.; Khoronenkova, S.V. Persistent DNA damage associated with ATM kinase deficiency promotes microglial dysfunction. Nucleic. Acids. Res. 2022, 50, 2700–2718. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Ma, F.; Herrup, K. Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J. Neurosci. 2019, 39, 6378–6394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinkle, J.T.; Patel, J.; Panicker, N.; Karuppagounder, S.S.; Biswas, D.; Belingon, B.; Chen, R.; Brahmachari, S.; Pletnikova, O.; Troncoso, J.C.; et al. STING mediates neurodegeneration and neuroinflammation in nigrostriatal α-synucleinopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2118819119. [Google Scholar] [CrossRef]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef]
- Giordano, A.M.S.; Luciani, M.; Gatto, F.; Alezz, M.A.; Beghè, C.; Della Volpe, L.; Migliara, A.; Valsoni, S.; Genua, M.; Dzieciatkowska, M.; et al. DNA damage contributes to neurotoxic inflammation in Aicardi-Goutières syndrome astrocytes. J. Exp. Med. 2022, 219, e20211121. [Google Scholar] [CrossRef]
- Song, X.; Ma, F.; Herrup, K. DNA damage-induced neuroinflammation in neurodegenerative disease. Alzheimer’s Dement. 2021, 17, e055175. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Welch, G.M.; Boix, C.A.; Schmauch, E.; Davila-Velderrain, J.; Victor, M.B.; Dileep, V.; Bozzelli, P.L.; Su, Q.; Cheng, J.D.; Lee, A.; et al. Neurons burdened by DNA double-strand breaks incite microglia activation through antiviral-like signaling in neurodegeneration. Sci. Adv. 2022, 8, eabo4662. [Google Scholar] [CrossRef]
- Sharma, M.; Rajendrarao, S.; Shahani, N.; Ramírez-Jarquín, U.N.; Subramaniam, S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc. Natl. Acad. Sci. USA 2022, 117, 15989–15999. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e618. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Erekat, N.S. Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Cambridge, UK, 2018. [Google Scholar]
- Luo, Q.; Sun, W.; Wang, Y.F.; Li, J.; Li, D.W. Association of p53 with Neurodegeneration in Parkinson’s Disease. Parkinsons. Dis. 2022, 2022, 6600944. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Zhu, X.; Ladenheim, B.; Yu, Q.-S.; Guo, Z.; Oyler, J.; Cutler, R.G.; Cadet, J.L.; Greig, N.H.; Mattson, M.P. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol. 2002, 52, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Davis, B.; Chiang, Y.-H.; Filichia, E.; Barnett, A.; Greig, N.H.; Hoffer, B.; Luo, Y. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson’s disease model. J. Neurochem. 2016, 138, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.J.; Huang, H.Y.; Chiu, T.L.; Chang, H.F.; Wu, H.R. Peroxiredoxin 5 Silencing Sensitizes Dopaminergic Neuronal Cells to Rotenone via DNA Damage-Triggered ATM/p53/PUMA Signaling-Mediated Apoptosis. Cells 2019, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Tiffany-Castiglioni, E. Paraquat-induced apoptosis in human neuroblastoma SH-SY5Y cells: Involvement of p53 and mitochondria. J. Toxicol. Environ. Health A 2008, 71, 289–299. [Google Scholar] [CrossRef]
- Nair, V.D. Activation of p53 signaling initiates apoptotic death in a cellular model of Parkinson’s disease. Apoptosis 2006, 11, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, J.; Ke, Y.; Zeng, X.; Gao, J.; Ba, X.; Wang, R. The key players of parthanatos: Opportunities for targeting multiple levels in the therapy of parthanatos-based pathogenesis. Cell Mol. Life Sci. 2022, 79, 60. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.-W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrabi, S.A.; Umanah, G.K.E.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagné, J.-P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.-W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kim, N.S.; Haince, J.-F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef] [Green Version]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef]
- Wang, Y.; An, R.; Umanah, G.K.; Park, H.; Nambiar, K.; Eacker, S.M.; Kim, B.; Bao, L.; Harraz, M.M.; Chang, C.; et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 2016, 354, aad6872. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP-1 and PARthanatos in ischemic stroke. J. Neurochem. 2022, 160, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chen, G.; Jin, W.; Mao, K.; Wan, H.; He, Y. Molecular Mechanisms of Parthanatos and Its Role in Diverse Diseases. Int. J. Mol. Sci. 2022, 23, 7292. [Google Scholar] [CrossRef]
- Lee, Y.; Karuppagounder, S.; Shin, J.-H.; Lee, Y.-I.; Ko, H.S.; Swing, D.; Jiang, H.; Kang, S.-U.; Lee, B.D.; Kang, H.C.; et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 2013, 16, 1392–1400. [Google Scholar] [CrossRef]
- Park, H.; Kam, T.-I.; Peng, H.; Chou, S.-C.; Mehrabani-Tabari, A.A.; Song, J.-J.; Yin, X.; Karuppagounder, S.S.; Umanah, G.K.; Rao, A.S.; et al. PAAN/MIF nuclease inhibition prevents neurodegeneration in Parkinson’s disease. Cell 2022, 185, 1943–1959.e1921. [Google Scholar] [CrossRef]
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.-Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Kodavati, M.; Britz, G.W.; Hegde, M.L. DNA Damage and Repair Deficiency in ALS/FTD-Associated Neurodegeneration: From Molecular Mechanisms to Therapeutic Implication. Front. Mol. Neurosci. 2021, 14, 784361. [Google Scholar] [CrossRef]
- Petr, M.A.; Tulika, T.; Carmona-Marin, L.M.; Scheibye-Knudsen, M. Protecting the Aging Genome. Trends Cell Biol. 2020, 30, 117–132. [Google Scholar] [CrossRef] [Green Version]
- Tuxworth, R.I.; Taylor, M.J.; Anduaga, A.M.; Hussien-Ali, A.; Chatzimatthaiou, S.; Longland, J.; Thompson, A.M.; Almutiri, S.; Alifragis, P.; Kyriacou, C.P.; et al. Attenuating the DNA damage response to double-strand breaks restores function in models of CNS neurodegeneration. Brain Commun. 2019, 1, fcz005. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K.; et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci. Transl. Med. 2014, 6, 268ra178. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Thompson, A.M.; Alhajlah, S.; Tuxworth, R.I.; Ahmed, Z. Inhibition of Chk2 promotes neuroprotection, axon regeneration, and functional recovery after CNS injury. Sci. Adv. 2022, 8, eabq2611. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Enforced DNA repair enzymes rescue neurons from apoptosis induced by target deprivation and axotomy in mouse models of neurodegeneration. Mech. Ageing. Dev. 2017, 161, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, W.P.; Hoeijmakers, J.H.; Pothof, J. Genome Integrity in Aging: Human Syndromes, Mouse Models, and Therapeutic Options. Annu. Rev. Pharmacol. Toxicol. 2022, 56, 427–445. [Google Scholar] [CrossRef]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Ruszkiewicz, J.A.; Bürkle, A.; Mangerich, A. Fueling genome maintenance: On the versatile roles of NAD(+) in preserving DNA integrity. J. Biol. Chem. 2022, 298, 102037. [Google Scholar] [CrossRef]
- Verdin, E. NAD⁺ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Nie, H.; Wei, X.; Fu, S.; Ying, W. NAD+ treatment can prevent rotenone-induced increases in DNA damage, Bax levels and nuclear translocation of apoptosis-inducing factor in differentiated PC12 cells. Neurochem. Res. 2015, 40, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Costa, A.C.; Celardo, I.; Loh, S.H.; Martins, L.M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 2016, 7, e2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brakedal, B.; Dölle, C.; Riemer, F.; Ma, Y.; Nido, G.S.; Skeie, G.O.; Craven, A.R.; Schwarzlmüller, T.; Brekke, N.; Diab, J.; et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022, 34, 396–407.e396. [Google Scholar] [CrossRef]
- Hong, Y.; Nie, H.; Wu, D.; Wei, X.; Ding, X.; Ying, W. NAD(+) treatment prevents rotenone-induced apoptosis and necrosis of differentiated PC12 cells. Neurosci. Lett. 2014, 560, 46–50. [Google Scholar] [CrossRef]
- Lu, L.; Tang, L.; Wei, W.; Hong, Y.; Chen, H.; Ying, W.; Chen, S. Nicotinamide mononucleotide improves energy activity and survival rate in an in vitro model of Parkinson’s disease. Exp. Ther. Med. 2014, 8, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Li, X.; Gao, H.; Feng, Z.; Li, X.; Zhao, L.; Jia, X.; Zhang, H.; Liu, J. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 2083–2090. [Google Scholar] [CrossRef]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open. 2017, 6, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Lagunas-Rangel, F.A. Current role of mammalian sirtuins in DNA repair. DNA Repair. 2019, 80, 85–92. [Google Scholar] [CrossRef]
- Yousafzai, N.A.; Zhou, Q.; Xu, W.; Shi, Q.; Xu, J.; Feng, L.; Chen, H.; Shin, V.Y.; Jin, H.; Wang, X. SIRT1 deacetylated and stabilized XRCC1 to promote chemoresistance in lung cancer. Cell Death Dis. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamori, T.; DeRicco, J.; Naqvi, A.; Hoffman, T.A.; Mattagajasingh, I.; Kasuno, K.; Jung, S.-B.; Kim, C.-S.; Irani, K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic. Acids. Res. 2010, 38, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Luo, J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Huh, Y.J.; Cho, H.-J.; Lee, B.; Park, J.; Hwang, D.-Y.; Kim, D.-W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem. Cell Rep. 2017, 9, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.-H.; Min, B.-H.; Lee, K.-M.; Cho, M.-H.; Park, G.-H.; Lee, K.-H. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Casta, A.; Wang, R.; Lozada, E.; Fan, W.; Kane, S.; Ge, Q.; Gu, W.; Orren, D.; Luo, J. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J. Biol. Chem. 2008, 283, 7590–7598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Head, P.E.; Daddacha, W.; Park, S.-H.; Li, X.; Pan, Y.; Madden, M.Z.; Duong, D.M.; Xie, M.; Yu, B.; et al. ATRIP Deacetylation by SIRT2 Drives ATR Checkpoint Activation by Promoting Binding to RPA-ssDNA. Cell Rep. 2016, 14, 1435–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, T.; Kagawa, W.; Ogi, T.; Kato, T.A.; Suzuki, T.; Dohmae, N.; Takizawa, K.; Nakazawa, Y.; Genet, M.D.; Saotome, M.; et al. Novel function of HATs and HDACs in homologous recombination through acetylation of human RAD52 at double-strand break sites. PLoS Genet. 2018, 14, e1007277. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [Green Version]
- Hwang, B.-J.; Jin, J.; Gao, Y.; Shi, G.; Madabushi, A.; Yan, A.; Guan, X.; Zalzman, M.; Nakajima, S.; Lan, L.; et al. SIRT6 protein deacetylase interacts with MYH DNA glycosylase, APE1 endonuclease, and Rad9-Rad1-Hus1 checkpoint clamp. BMC. Mol. Biol. 2015, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L.; et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, J.A.; Ghirotto, B.; Targhetta, V.P.; de Lima, J.; Câmara, N.O.S. Sirtuins as pharmacological targets in neurodegenerative and neuropsychiatric disorders. Br. J. Pharmacol. 2022, 179, 1496–1511. [Google Scholar] [CrossRef]
- Li, X.; Feng, Y.; Wang, X.X.; Truong, D.; Wu, Y.C. The Critical Role of SIRT1 in Parkinson’s Disease: Mechanism and Therapeutic Considerations. Aging Dis. 2020, 11, 1608–1622. [Google Scholar] [CrossRef]
- Zhang, L.-F.; Yu, X.-L.; Ji, M.; Liu, S.-Y.; Wu, X.-L.; Wang, Y.-J.; Liu, R.-T. Resveratrol alleviates motor and cognitive deficits and neuropathology in the A53T α-synuclein mouse model of Parkinson’s disease. Food. Funct. 2018, 9, 6414–6426. [Google Scholar] [CrossRef]
- Su, C.F.; Jiang, L.; Zhang, X.W.; Iyaswamy, A.; Li, M. Resveratrol in Rodent Models of Parkinson’s Disease: A Systematic Review of Experimental Studies. Front. Pharmacol. 2021, 12, 644219. [Google Scholar] [CrossRef]
- Chen, H.H.; Chang, P.C.; Chen, C.; Chan, M.H. Protective and therapeutic activity of honokiol in reversing motor deficits and neuronal degeneration in the mouse model of Parkinson’s disease. Pharmacol. Rep. 2018, 70, 668–676. [Google Scholar] [CrossRef]
- Merlo, D.; Mollinari, C.; Racaniello, M.; Garaci, E.; Cardinale, A. DNA Double Strand Breaks: A Common Theme in Neurodegenerative Diseases. Curr. Alzheimer. Res. 2016, 13, 1208–1218. [Google Scholar] [CrossRef]
- Kruman, I.I.; Wersto, R.P.; Cardozo-Pelaez, F.; Smilenov, L.; Chan, S.L.; Chrest, F.J.; Emokpae, R., Jr.; Gorospe, M.; Mattson, M.P. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 2004, 41, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Schepici, G.; Silvestro, S.; Bramanti, P.; Mazzon, E. Caffeine: An Overview of Its Beneficial Effects in Experimental Models and Clinical Trials of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 4766. [Google Scholar] [CrossRef]
- Aditi; Downing, S.M.; Schreiner, P.A.; Kwak, Y.D.; Li, Y.; Shaw, T.I.; Russell, H.R.; McKinnon, P.J. Genome instability independent of type I interferon signaling drives neuropathology caused by impaired ribonucleotide excision repair. Neuron 2021, 109, 3962–3979.e3966. [Google Scholar] [CrossRef] [PubMed]
- Maor-Nof, M.; Shipony, Z.; Lopez-Gonzalez, R.; Nakayama, L.; Zhang, Y.-J.; Couthouis, J.; Blum, J.A.; Castruita, P.A.; Linares, G.R.; Ruan, K.; et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 2021, 184, 689–708.e620. [Google Scholar] [CrossRef]
- Liang, Z.-Q.; Li, Y.-L.; Zhao, X.-L.; Han, R.; Wang, X.-X.; Wang, Y.; Chase, T.N.; Bennett, M.C.; Qin, Z.-H. NF-kappaB contributes to 6-hydroxydopamine-induced apoptosis of nigral dopaminergic neurons through p53. Brain Res. 2007, 1145, 190–203. [Google Scholar] [CrossRef]
- Mendrysa, S.M.; Ghassemifar, S.; Malek, R. p53 in the CNS: Perspectives on Development, Stem Cells, and Cancer. Genes Cancer 2011, 2, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, P.; Frost, B.; Peng, S.; Yang, Y.J.; Park, P.J.; Feany, M. p53 prevents neurodegeneration by regulating synaptic genes. Proc. Natl. Acad. Sci. USA 2014, 111, 18055–18060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Jenner, P.; Przedborski, S. Pathogenesis of Parkinson’s disease. Mov. Disord. 2013, 28, 24–30. [Google Scholar] [CrossRef]
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The cell biology of Parkinson’s disease. J. Cell Biol. 2021, 220, e202012095. [Google Scholar] [CrossRef]
- Chow, H.M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef]
- McKinnon, P.J. Maintaining genome stability in the nervous system. Nat. Neurosci. 2013, 16, 1523–1529. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Target | Drugs | Models | Effects | References |
|---|---|---|---|---|
| NAD | NMN | Rotenone-treated PC12 cells | DNA damage ↓; energy metabolism ↑; apoptosis ↓; necrosis ↓. | [183,187,188] |
| NAM | MPP+-treated SK-N-MC cells; α-synuclein transgenic drosophila | Mitochondrial function ↑; oxidative stress ↓; DNA damage ↓; motor function ↑. | [189] | |
| Parkin or Pink1 mutant drosophila | Mitochondrial function ↑; loss of dopaminergic neurons ↓. | [185,190] | ||
| NR | GBA-PD iPSC neurons and fly models of PD | Mitochondrial function ↑; autophagy ↑; motor function ↑. | [184] | |
| Double-blinded phase I clinical in PD patients | Mitochondrial function ↑; lysosomal and proteasomal function ↑; inflammation ↓; | [186] | ||
| SIRT1 | Resveratrol | MPTP/6-ODHA/Rotenone-induced PD rodent models; A53T α-synuclein mice | α-synuclein pathology ↓; TH ↑; inflammation ↓; mitochondrial function ↑; oxidative stress ↓; motor function ↑ | [207,208] |
| SIRT3 | Honokiol | 6-OHDA-lesioned mice | Oxidative stress ↓; TH ↑; motor function ↑ | [209] |
| PARP1 | Veliparib Rucaparib Talazopari | α-synuclein PFF-treated primary neurons and mice | α-synuclein pathology ↓; TH ↑; parthanatos ↓; motor function ↑ | [19] |
| ATM | KU-55933 | MPP+-treated cerebellar granule cells | DNA damage ↓; apoptosis ↓ | [36] |
| Caffeine | 6-OHDA-treated PC12 cells | DNA damage ↓; apoptosis ↓ | [152] | |
| p53 | Pifithrin-α | Paraquat-treated SY5Y cells | Mitochondrial function ↑; DNA damage ↓; apoptosis ↓ | [151] |
| Z-1-117 | MPTP-treated mice | TH ↑; apoptosis ↓; motor function ↑ | [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.-X.; Li, Y.-L.; Pu, J.-L.; Zhang, B.-R. DNA Damage-Mediated Neurotoxicity in Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 6313. https://doi.org/10.3390/ijms24076313
Wang Z-X, Li Y-L, Pu J-L, Zhang B-R. DNA Damage-Mediated Neurotoxicity in Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(7):6313. https://doi.org/10.3390/ijms24076313
Chicago/Turabian StyleWang, Zhong-Xuan, Yao-Lin Li, Jia-Li Pu, and Bao-Rong Zhang. 2023. "DNA Damage-Mediated Neurotoxicity in Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 7: 6313. https://doi.org/10.3390/ijms24076313