
Lanthanides as Calcium Mimetic Species in Calcium-Signaling/Buffering Proteins: The Effect of Lanthanide Type on the Ca2+/Ln3+ Competition


Abstract
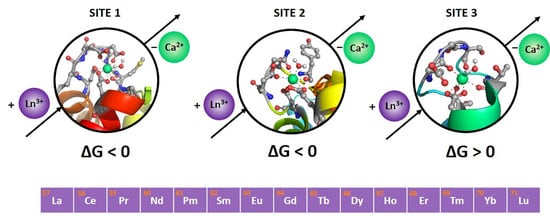
:
1. Introduction
2. Results and Discussion
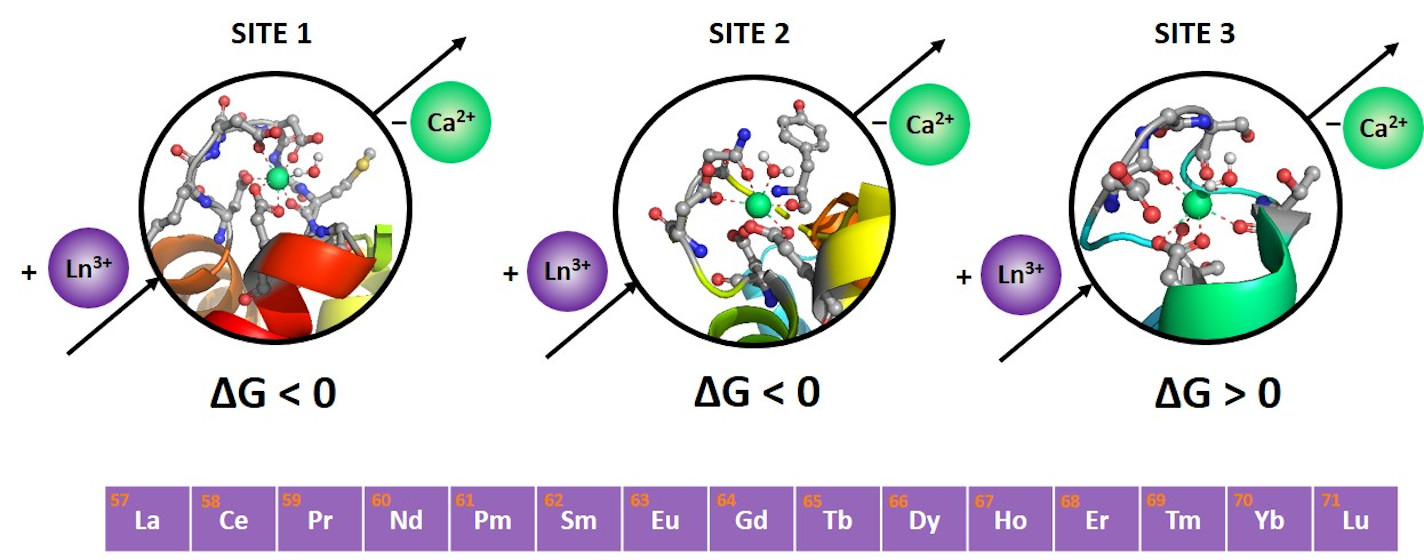
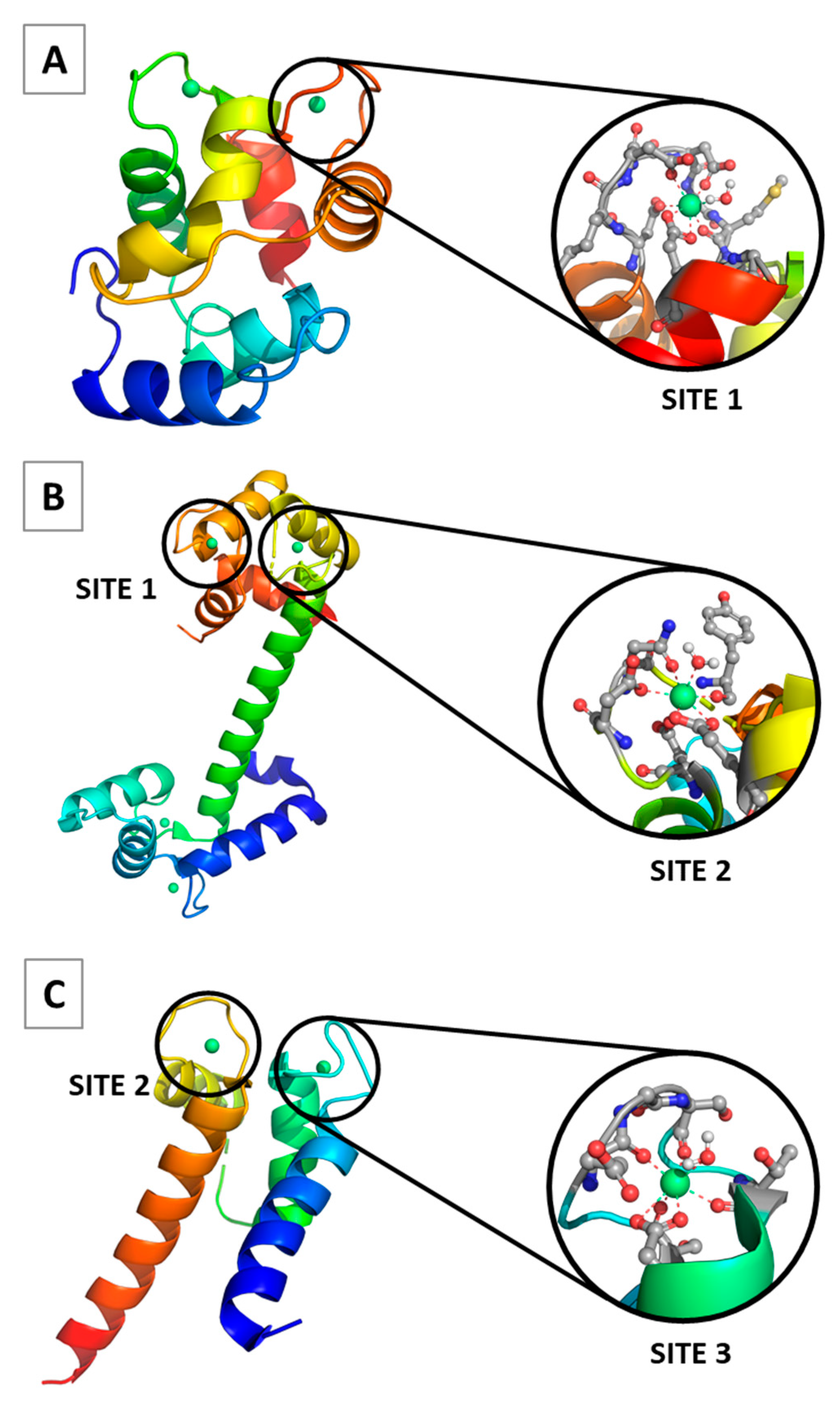
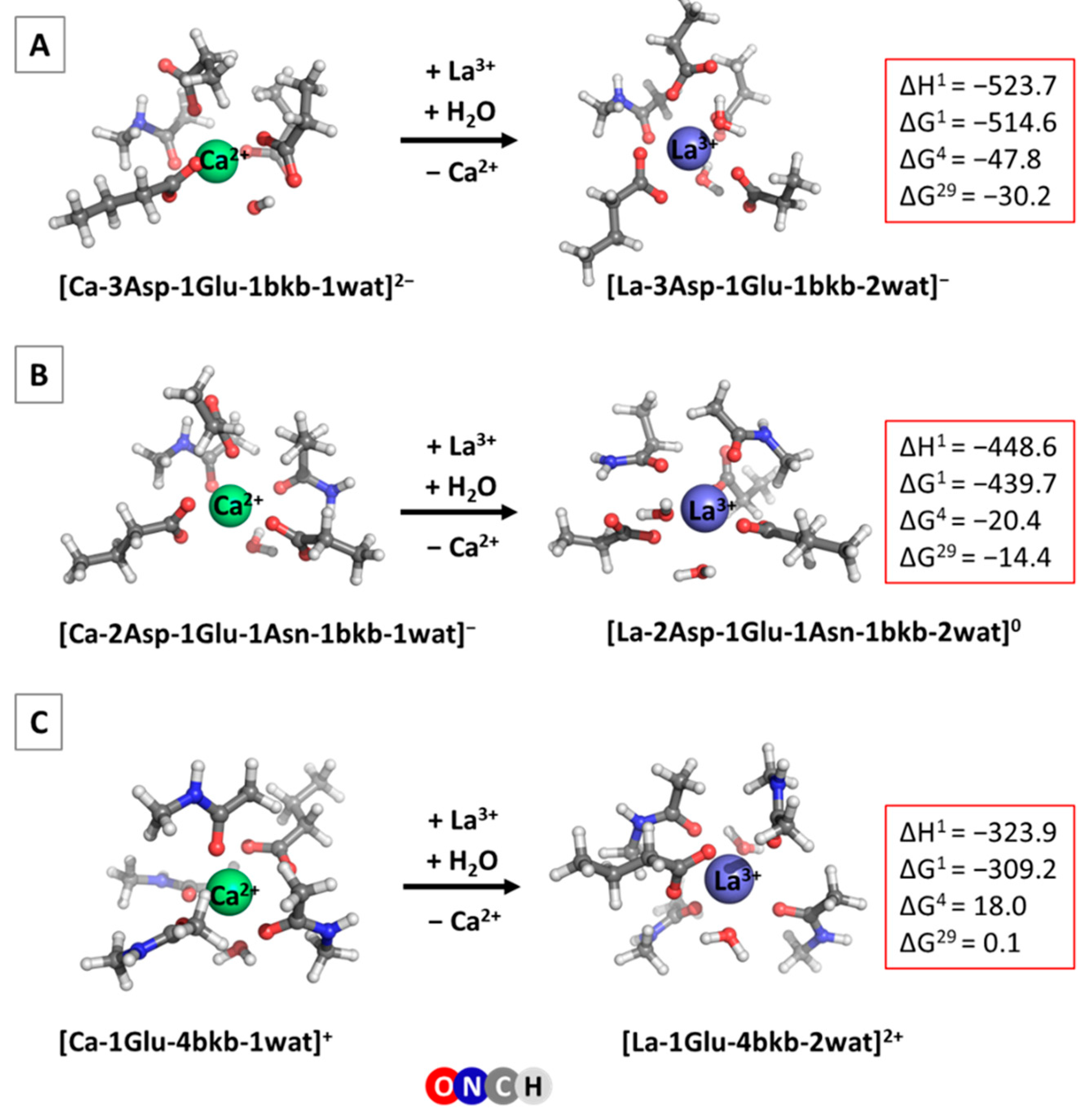
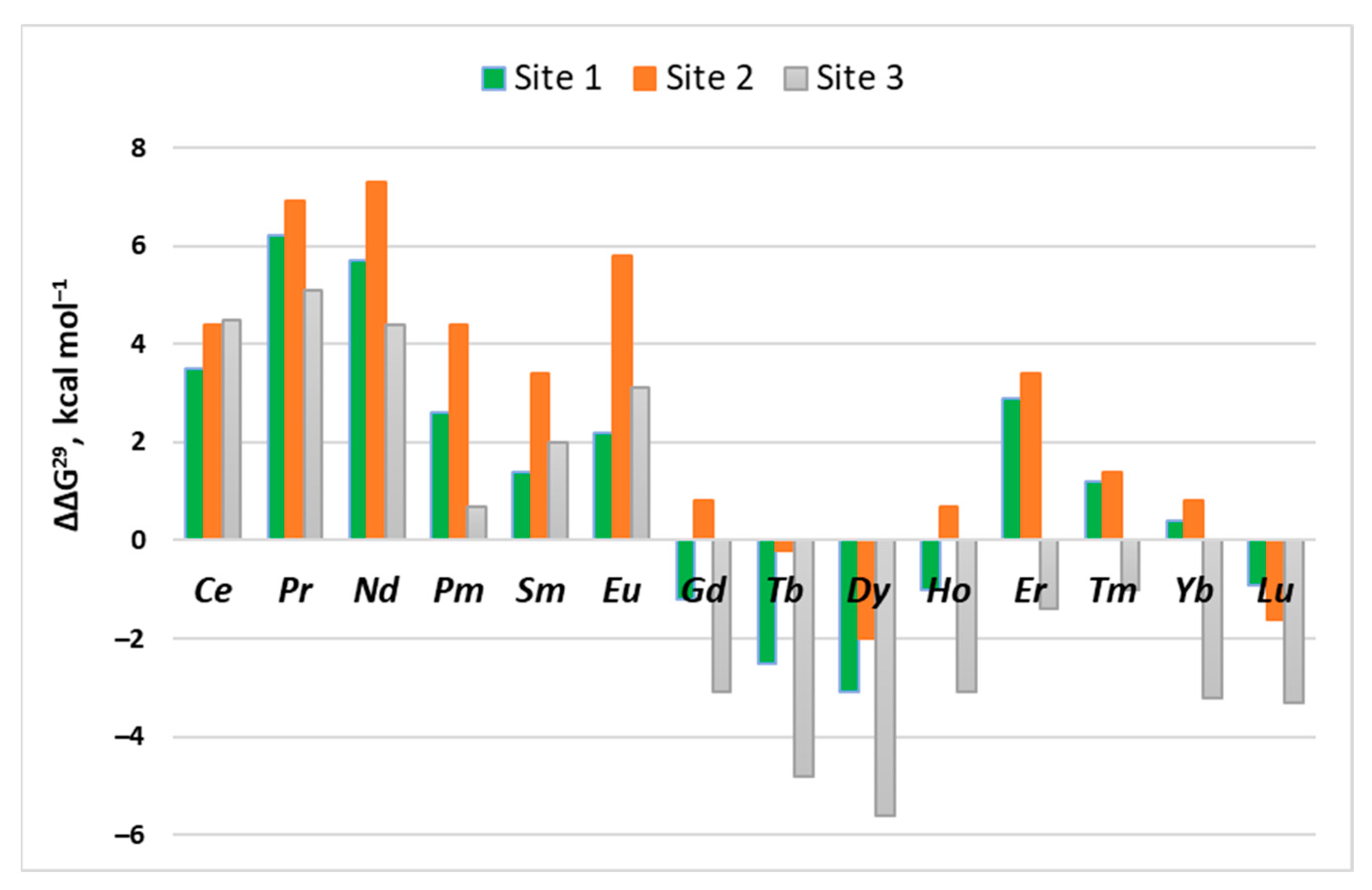
2.1. Site 1
2.2. Site 2
2.3. Site 3
3. Materials and Methods
3.1. Models Used
3.2. DFT/PCM Calculations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
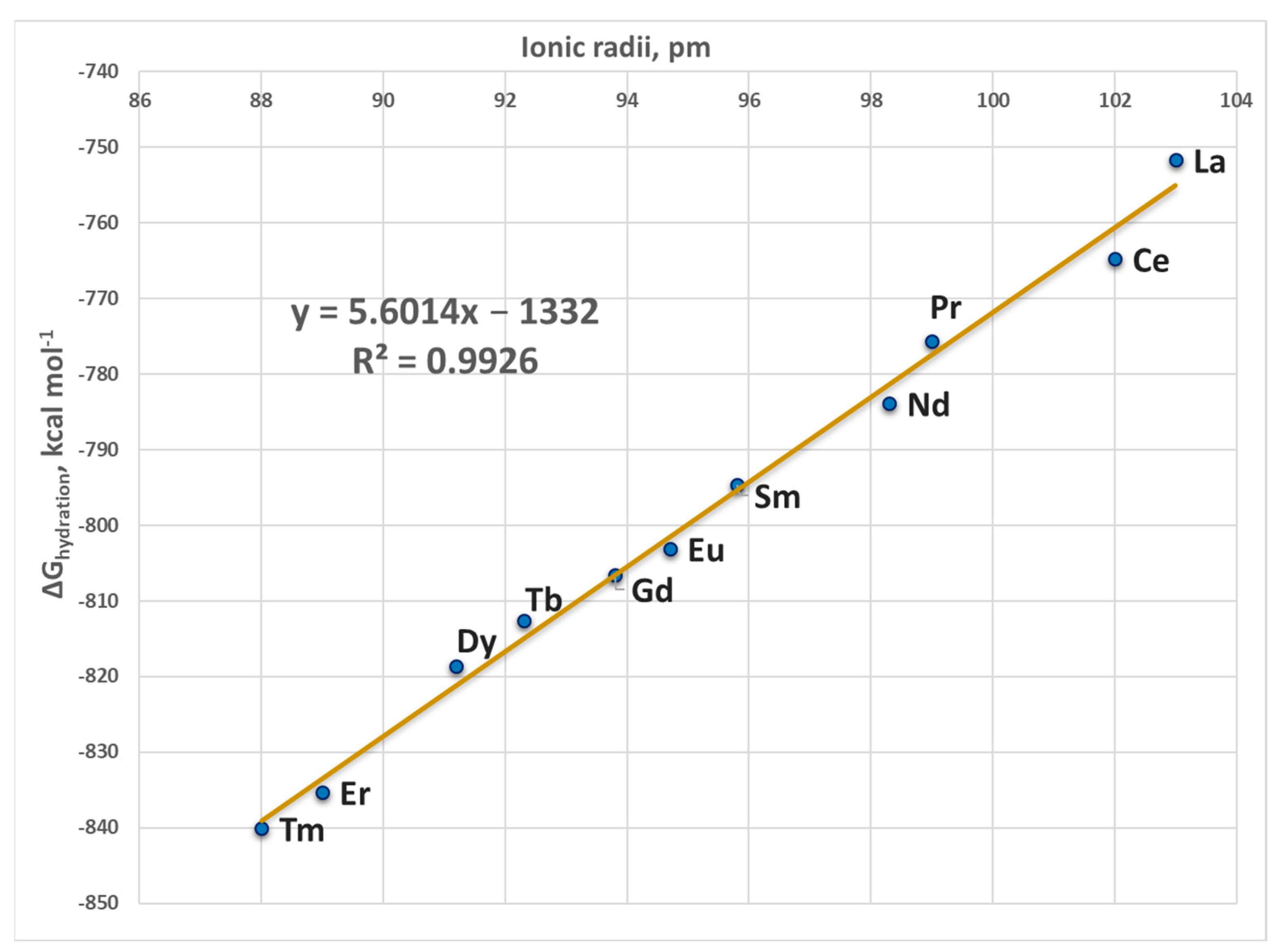
| Metal Cation | Ionic Radius a (Å) | ΔGhydration (kcal mol−1) b | ECP |
|---|---|---|---|
| La3+ | 1.03 | −751.7 | MWB46 |
| Ce3+ | 1.02 | −764.8 | MWB47 |
| Pr3+ | 0.99 | −775.6 | MWB48 |
| Nd3+ | 0.983 | −783.9 | MWB49 |
| Pm3+ | 0.970 | −788.7 c | MWB50 |
| Sm3+ | 0.958 | −794.7 | MWB51 |
| Eu3+ | 0.947 | −803.1 | MWB52 |
| Gd3+ | 0.938 | −806.6 | MWB53 |
| Tb3+ | 0.923 | −812.6 | MWB54 |
| Dy3+ | 0.912 | −818.6 | MWB55 |
| Ho3+ | 0.901 | −827.3 c | MWB56 |
| Er3+ | 0.89 | −835.3 | MWB57 |
| Tm3+ | 0.88 | −840.1 | MWB58 |
| Yb3+ | 0.868 | −845.8 c | MWB59 |
| Lu3+ | 0.861 | −849.7 c | MWB60 |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bruno, J.A.; Horrocks, W.D.W.; Zauhar, R.J. Europium(III) Luminescence and Tyrosine to Terbium(III) Energy-Transfer Studies of Invertebrate (Octopus) Calmodulin. Biochemistry 1992, 31, 7016–7026. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.A.; Leavis, P.C.; Horrocks, W.D.W.; Gergely, J. Binding of Lanthanide Ions to Troponin C. Biochemistry 1981, 20, 2439–2444. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Horrocks, W.D.; Amburgey, J.C.; Weber, D.J. Characterization of Lanthanide Ion Binding to the EF-Hand Protein S100 Beta by Luminescence Spectroscopy. Biochemistry 1997, 36, 9674–9680. [Google Scholar] [CrossRef]
- Drakenberg, T.; Swärd, M.; Cavé, A.; Parello, J. Metal-Ion Binding to Parvalbumin. A 113Cd-n.m.r. Study of the Binding of Different Lanthanide Ions. Biochem. J. 1985, 227, 711. [Google Scholar] [CrossRef] [Green Version]
- Edington, S.C.; Gonzalez, A.; Middendorf, T.R.; Halling, D.B.; Aldrich, R.W.; Baiz, C.R. Coordination to Lanthanide Ions Distorts Binding Site Conformation in Calmodulin. Proc. Natl. Acad. Sci. USA 2018, 115, E3126–E3134. [Google Scholar] [CrossRef] [Green Version]
- Bertini, I.; Gelis, I.; Katsaros, N.; Luchinat, C.; Provenzani, A. Tuning the Affinity for Lanthanides of Calcium Binding Proteins. Biochemistry 2003, 42, 8011–8021. [Google Scholar] [CrossRef]
- Barbieri, R.; Bertini, I.; Cavallaro, G.; Lee, Y.M.; Luchinat, C.; Rosato, A. Paramagnetically Induced Residual Dipolar Couplings for Solution Structure Determination of Lanthanide Binding Proteins. J. Am. Chem. Soc. 2002, 124, 5581–5587. [Google Scholar] [CrossRef] [PubMed]
- Mulqueen, P.; Tingey, J.M.; Horrocks, W.D.W. Characterization of Lanthanide(III) Ion Binding to Calmodulin Using Luminescence Spectroscopy. Biochemistry 1985, 24, 6639–6645. [Google Scholar] [CrossRef]
- Buccigross, J.M.; Nelson, D.J. EPR Studies Show That All Lanthanides Do Not Have the Same Order of Binding to Calmodulin. Biochem. Biophys. Res. Commun. 1986, 138, 1243–1249. [Google Scholar] [CrossRef]
- Rhee, M.-J.; Sudnick, D.R.; Arkle, V.K.; Horrocks, W.D. Lanthanide Ion Luminescence Probes. Characterization of Metal Ion Binding Sites and Intermetal Energy Transfer Distance Measurements in Calcium-Binding Proteins. 1. Parvalbumin. Biochemistry 1981, 20, 3328–3334. [Google Scholar] [CrossRef]
- Lee, L.; Sykes, B.D. Strategies for the Uses of Lanthanide NMR Shift Probes in the Determination of Protein Structure in Solutio. Application to the EF Calcium Binding Site of Carp Parvalbumin. Biophys. J. 1980, 32, 193–210. [Google Scholar] [CrossRef] [Green Version]
- Kupke, D.W.; Fox, J.W. Volume Changes in the Binding of Lanthanides to Peptide Analogues of Loop II of Calmodulin. Biochemistry 1989, 28, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Donaire, A.; Jiménez, B.; Luchinat, C.; Parigi, G.; Piccioli, M.; Poggi, L. Paramagnetism-Based versus Classical Constraints: An Analysis of the Solution Structure of Ca Ln Calbindin D9k. J. Biomol. NMR 2001, 21, 85–98. [Google Scholar] [CrossRef]
- Cotruvo, J.A.J. The Chemistry of Lanthanides in Biology: Recent Discoveries, Emerging Principles, and Technological Applications. ACS Cent. Sci. 2019, 5, 1496–1506. [Google Scholar] [CrossRef] [Green Version]
- Mattocks, J.A.; Cotruvo, J.A.; Deblonde, G.J.-P. Engineering Lanmodulin’s Selectivity for Actinides over Lanthanides by Controlling Solvent Coordination and Second-Sphere Interactions. Chem. Sci. 2022, 13, 6054–6066. [Google Scholar] [CrossRef]
- Mattocks, J.A.; Tirsch, J.L.; Cotruvo, J.A. Chapter Two—Determination of Affinities of Lanthanide-Binding Proteins Using Chelator-Buffered Titrations. In Rare-Earth Element Biochemistry: Characterization and Applications of Lanthanide-Binding Biomolecules; Cotruvo, J.A., Jr., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 651, pp. 23–61. ISBN 0076-6879. [Google Scholar]
- Caldwell, S.J.; Haydon, I.C.; Piperidou, N.; Huang, P.-S.; Bick, M.J.; Sjöström, H.S.; Hilvert, D.; Baker, D.; Zeymer, C. Tight and Specific Lanthanide Binding in a de Novo TIM Barrel with a Large Internal Cavity Designed by Symmetric Domain Fusion. Proc. Natl. Acad. Sci. USA 2020, 117, 30362–30369. [Google Scholar] [CrossRef]
- Takacs, M. Adapting Proteins for Clinical and Industrial Use. Ph.D. Thesis, Syracuse University, Syracuse, NY, USA, 2018. [Google Scholar]
- Dudev, T.; Chang, L.-Y.; Lim, C. Factors Governing the Substitution of La3+ for Ca2+ and Mg2+ in Metalloproteins: A DFT/CDM Study. J. Am. Chem. Soc. 2005, 127, 4091–4103. [Google Scholar] [CrossRef] [PubMed]
- Cates, M.S.; Berry, M.B.; Ho, E.L.; Li, Q.; Potter, J.D.; Phillips, G.N. Metal-Ion Affinity and Specificity in EF-Hand Proteins: Coordination Geometry and Domain Plasticity in Parvalbumin. Structure 1999, 7, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Gifford, J.L.; Walsh, M.P.; Vogel, H.J. Structures and Metal-Ion-Binding Properties of the Ca2+-Binding Helix-Loop-Helix EF-Hand Motifs. Biochem. J. 2007, 405, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Declercq, J.P.; Tinant, B.; Parello, J.; Rambaud, J. Ionic Interactions with Parvalbumins: Crystal Structure Determination of Pike 4.10 Parvalbumin in Four Different Ionic Environments. J. Mol. Biol. 1991, 220, 1017–1039. [Google Scholar] [CrossRef]
- Sarhan, M.F.; Tung, C.C.; Van Petegem, F.; Ahern, C.A. Crystallographic Basis for Calcium Regulation of Sodium Channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3558–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, G.; Ding, Y.; Wang, Z.; Barraclough, R.; Rudland, P.S.; Fernig, D.G.; Rao, Z. The Crystal Structure at 2 Å Resolution of the Ca2+-Binding Protein S100P. J. Mol. Biol. 2003, 325, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; Wilkinson, G. Advanced Inorganic Chemistry, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1988. [Google Scholar]
- Evans, C.H. Biochemistry of the Lanthanides; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Gschneidner, K.A.; Eyring, L.; Gschneidner, K.A. Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Eyring, L., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 15. [Google Scholar]
- Gschneidner, K.A.; Eyring, L.; Lander, G.H.; Choppin, G.R. Handbook on the Physics and Chemistry of Rare Earths; Elsevier: Amsterdam, The Netherlands, 1994; Volume 18. [Google Scholar]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Dean, J.A. Lange’ s Handbook of Chemistry; McGraw-Hill, Inc.: New York, NY, USA, 1985. [Google Scholar]
- Abola, E.E.; Sussman, J.L.; Prilusky, J.; Manning, N.O. Protein Data Bank Archives of Three-Dimensional Macromolecular Structures. Methods Enzymol. 1997, 277, 556–571. [Google Scholar] [CrossRef]
- Pidcock, E.; Moore, G.R. Structural Characteristics of Protein Binding Sites for Calcium and Lanthanide Ions. J. Biol. Inorg. Chem. 2001, 6, 479–489. [Google Scholar] [CrossRef]
- Nikolova, V.; Angelova, S.; Markova, N.; Dudev, T. Gallium as a Therapeutic Agent: A Thermodynamic Evaluation of the Competition between Ga3+ and Fe3+ Ions in Metalloproteins. J. Phys. Chem. B 2016, 120, 2241–2248. [Google Scholar] [CrossRef]
- Dudev, T.; Mazmanian, K.; Lim, C. Factors Controlling the Selectivity for Na+ over Mg2+ in Sodium Transporters and Enzymes. Phys. Chem. Chem. Phys. 2016, 18, 16986–16997. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. Competition between Li+ and Mg2+ in Metalloproteins. Implications for Lithium Therapy. J. Am. Chem. Soc. 2011, 133, 9506–9515. [Google Scholar] [CrossRef]
- Dudev, T.; Musset, B.; Morgan, D.; Cherny, V.V.; Smith, S.M.E.; Mazmanian, K.; DeCoursey, T.E.; Lim, C. Selectivity Mechanism of the Voltage-Gated Proton Channel, HV1. Sci. Rep. 2015, 5, 10320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudev, T.; Lim, C. Ion Selectivity in the Selectivity Filters of Acid-Sensing Ion Channels. Sci. Rep. 2015, 5, 7864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudev, T.; Lim, C. Importance of Metal Hydration on the Selectivity of Mg2+ versus Ca2+ in Magnesium Ion Channels. J. Am. Chem. Soc. 2013, 135, 17200–17208. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. The Effect of Metal Binding on the Characteristic Infrared Band Intensities of Ligands of Biological Interest. J. Mol. Struct. 2012, 1009, 83–88. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Ion Selectivity Strategies of Sodium Channel Selectivity Filters. Acc. Chem. Res. 2014, 47, 3580–3587. [Google Scholar] [CrossRef] [PubMed]
- Kuppuraj, G.; Dudev, M.; Lim, C. Factors Governing Metal−Ligand Distances and Coordination Geometries of Metal Complexes. J. Phys. Chem. B 2009, 113, 2952–2960. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. Factors Governing the Na+ vs K+ Selectivity in Sodium Ion Channels. J. Am. Chem. Soc. 2010, 132, 2321–2332. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Competition among Ca2+, Mg2+, and Na+ for Model Ion Channel Selectivity Filters: Determinants of Ion Selectivity. J. Phys. Chem. B 2012, 116, 10703–10714. [Google Scholar] [CrossRef]
- Marcus, Y. Thermodynamics of Solvation of Ions. Part 5.—Gibbs Free Energy of Hydration at 298.15 K. J. Chem. Soc. Faraday Trans. 1991, 87, 2995–2999. [Google Scholar] [CrossRef]
- Kumar, R.P.; Ranaghan, M.J.; Ganjei, A.Y.; Oprian, D.D. Crystal Structure of Recoverin with Calcium Ions Bound to Both Functional EF Hands. Biochemistry 2015, 54, 7222–7228. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Dudev, T.; Cheshmedzhieva, D.; Doudeva, L. Competition between Abiogenic Al3+ and Native Mg2+, Fe2+ and Zn2+ Ions in Protein Binding Sites: Implications for Aluminum Toxicity. J. Mol. Model. 2018, 24, 55. [Google Scholar] [CrossRef]
- Dudev, T.; Nikolova, V. Determinants of Fe2+ over M2+ (M = Mg, Mn, Zn) Selectivity in Non-Heme Iron Proteins. Inorg. Chem. 2016, 55, 12644–12650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrev, S.; Kircheva, N.; Nikolova, V.; Angelova, S.; Dudev, T. Competition between Ag+ and Ni2+ in Nickel Enzymes: Implications for the Ag+ Antibacterial Activity. Comput. Biol. Chem. 2022, 101, 107785. [Google Scholar] [CrossRef] [PubMed]
- Kircheva, N.; Dobrev, S.; Nikolova, V.; Angelova, S.; Dudev, T. Theoretical Insight into the Phosphate-Targeted Silver’s Antibacterial Action: Differentiation between Gram (+) and Gram (−) Bacteria. Inorg. Chem. 2022, 61, 10089–10100. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Rev. D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Dudev, T.; Lim, C. Determinants of K+ vs Na+ Selectivity in Potassium Channels. J. Am. Chem. Soc. 2009, 131, 8092–8101. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nairn, A.; Marcus, Y. Solvation Thermodynamics of Nonionic Solutes. J. Chem. Phys. 1998, 81, 2016. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann: Oxford, UK; Sophia Forster & Andreas Frewer: Erlangen, Germany, 1997. [Google Scholar]




| Metal | Ca2+ → Ln3+ | La3+ → Ln3+ | ||||
|---|---|---|---|---|---|---|
| ΔG1 | ΔG4 | ΔG29 | ΔΔG1 | ΔΔG4 | ΔΔG29 | |
| La3+ | −514.6 | −47.8 | −30.2 | 0.0 | 0.0 | 0.0 |
| Ce3+ | −524.6 | −44.4 | −26.7 | −10.0 | 3.4 | 3.5 |
| Pr3+ | −532.6 | −41.6 | −24.0 | −18.0 | 6.2 | 6.2 |
| Nd3+ | −541.7 | −42.1 | −24.5 | −26.7 | 5.7 | 5.7 |
| Pm3+ | −549.1 | −45.2 | −27.6 | −34.5 | 2.6 | 2.6 |
| Sm3+ | −556.7 | −46.3 | −28.8 | −41.7 | 1.5 | 1.4 |
| Eu3+ | −563.8 | −45.4 | −28.0 | −49.2 | 2.4 | 2.2 |
| Gd3+ | −570.7 | −48.8 | −31.4 | −56.1 | −1.0 | −1.2 |
| Tb3+ | −577.8 | −50.1 | −32.7 | −63.4 | −2.3 | −2.5 |
| Dy3+ | −584.6 | −50.7 | −33.3 | −70.0 | −2.9 | −3.1 |
| Ho3+ | −591.1 | −48.5 | −31.2 | −76.5 | −0.7 | −1.0 |
| Er3+ | −594.9 | −44.4 | −27.3 | −80.3 | 3.4 | 2.9 |
| Tm3+ | −601.3 | −46.0 | −29.0 | −86.7 | 1.8 | 1.2 |
| Yb3+ | −607.8 | −46.8 | −29.8 | −93.2 | 1.0 | 0.4 |
| Lu3+ | −613.0 | −48.1 | −31.1 | −98.4 | −0.3 | −0.9 |
| Metal | Ca2+ → Ln3+ | La3+ → Ln3+ | ||||
|---|---|---|---|---|---|---|
| ΔG1 | ΔG4 | ΔG29 | ΔΔG1 | ΔΔG4 | ΔΔG29 | |
| La3+ | −439.7 | −20.4 | −14.4 | 0.0 | 0.0 | 0.0 |
| Ce3+ | −448.7 | −16.1 | −10.0 | −9.0 | 4.3 | 4.4 |
| Pr3+ | −457.3 | −13.7 | −7.5 | −17.6 | 6.7 | 6.9 |
| Nd3+ | −464.9 | −13.1 | −7.1 | −25.2 | 7.3 | 7.3 |
| Pm3+ | −472.7 | −15.8 | −10.0 | −33.0 | 4.6 | 4.4 |
| Sm3+ | −480.4 | −17.1 | −11.0 | −40.7 | 3.3 | 3.4 |
| Eu3+ | −485.5 | −14.7 | −8.6 | −45.8 | 5.7 | 5.8 |
| Gd3+ | −494.9 | −19.8 | −13.6 | −55.2 | 0.6 | 0.8 |
| Tb3+ | −501.7 | −20.8 | −14.6 | −62.0 | −0.4 | −0.2 |
| Dy3+ | −509.5 | −22.5 | −16.4 | −69.8 | −2.1 | −2.0 |
| Ho3+ | −515.3 | −19.6 | −13.7 | −75.6 | 0.8 | 0.7 |
| Er3+ | −520.9 | −16.8 | −11.0 | −81.2 | 3.6 | 3.4 |
| Tm3+ | −527.3 | −19.1 | −13.0 | −87.6 | 1.3 | 1.4 |
| Yb3+ | −534.1 | −19.7 | −13.6 | −94.4 | 0.7 | 0.8 |
| Lu3+ | −539.9 | −22.1 | −16.0 | −100.2 | −1.7 | −1.6 |
| Metal | Ca2+ → Ln3+ | La3+ → Ln3+ | ||||
|---|---|---|---|---|---|---|
| ΔG1 | ΔG4 | ΔG29 | ΔΔG1 | ΔΔG4 | ΔΔG29 | |
| La3+ | −309.2 | 18.0 | 0.1 | 0.0 | 0.0 | 0.0 |
| Ce3+ | −316.9 | 21.2 | 4.6 | −7.7 | 3.2 | 4.5 |
| Pr3+ | −327.6 | 23.3 | 5.2 | −18.4 | 5.3 | 5.1 |
| Nd3+ | −336.4 | 22.8 | 4.5 | −27.2 | 4.8 | 4.4 |
| Pm3+ | −273.7 | 19.2 | 0.8 | −35.5 | 1.2 | 0.7 |
| Sm3+ | −349.0 | 20.6 | 2.1 | −39.8 | 2.6 | 2.0 |
| Eu3+ | −356.1 | 21.8 | 3.2 | −46.9 | 3.8 | 3.1 |
| Gd3+ | −365.7 | 15.7 | −3.0 | −56.5 | −2.3 | −3.1 |
| Tb3+ | −373.2 | 14.1 | −4.7 | −64.0 | −3.9 | −4.8 |
| Dy3+ | −379.6 | 13.4 | −5.5 | −70.4 | −4.6 | −5.6 |
| Ho3+ | −385.6 | 16.0 | −3.0 | −76.4 | −2.0 | −3.1 |
| Er3+ | −392.0 | 17.6 | −1.3 | −82.8 | −0.4 | −1.4 |
| Tm3+ | −396.2 | 18.1 | −0.9 | −87.0 | 0.1 | −1.0 |
| Yb3+ | −403.9 | 15.9 | −3.1 | −94.7 | −2.1 | −3.2 |
| Lu3+ | −407.8 | 15.8 | −3.2 | −98.6 | −2.2 | −3.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolova, V.; Kircheva, N.; Dobrev, S.; Angelova, S.; Dudev, T. Lanthanides as Calcium Mimetic Species in Calcium-Signaling/Buffering Proteins: The Effect of Lanthanide Type on the Ca2+/Ln3+ Competition. Int. J. Mol. Sci. 2023, 24, 6297. https://doi.org/10.3390/ijms24076297
Nikolova V, Kircheva N, Dobrev S, Angelova S, Dudev T. Lanthanides as Calcium Mimetic Species in Calcium-Signaling/Buffering Proteins: The Effect of Lanthanide Type on the Ca2+/Ln3+ Competition. International Journal of Molecular Sciences. 2023; 24(7):6297. https://doi.org/10.3390/ijms24076297
Chicago/Turabian StyleNikolova, Valya, Nikoleta Kircheva, Stefan Dobrev, Silvia Angelova, and Todor Dudev. 2023. "Lanthanides as Calcium Mimetic Species in Calcium-Signaling/Buffering Proteins: The Effect of Lanthanide Type on the Ca2+/Ln3+ Competition" International Journal of Molecular Sciences 24, no. 7: 6297. https://doi.org/10.3390/ijms24076297