
Transcriptome and GWAS Analyses Reveal Candidate Gene for Root Traits of Alfalfa during Germination under Salt Stress
 , , and
, , and
Abstract
:1. Introduction
2. Results
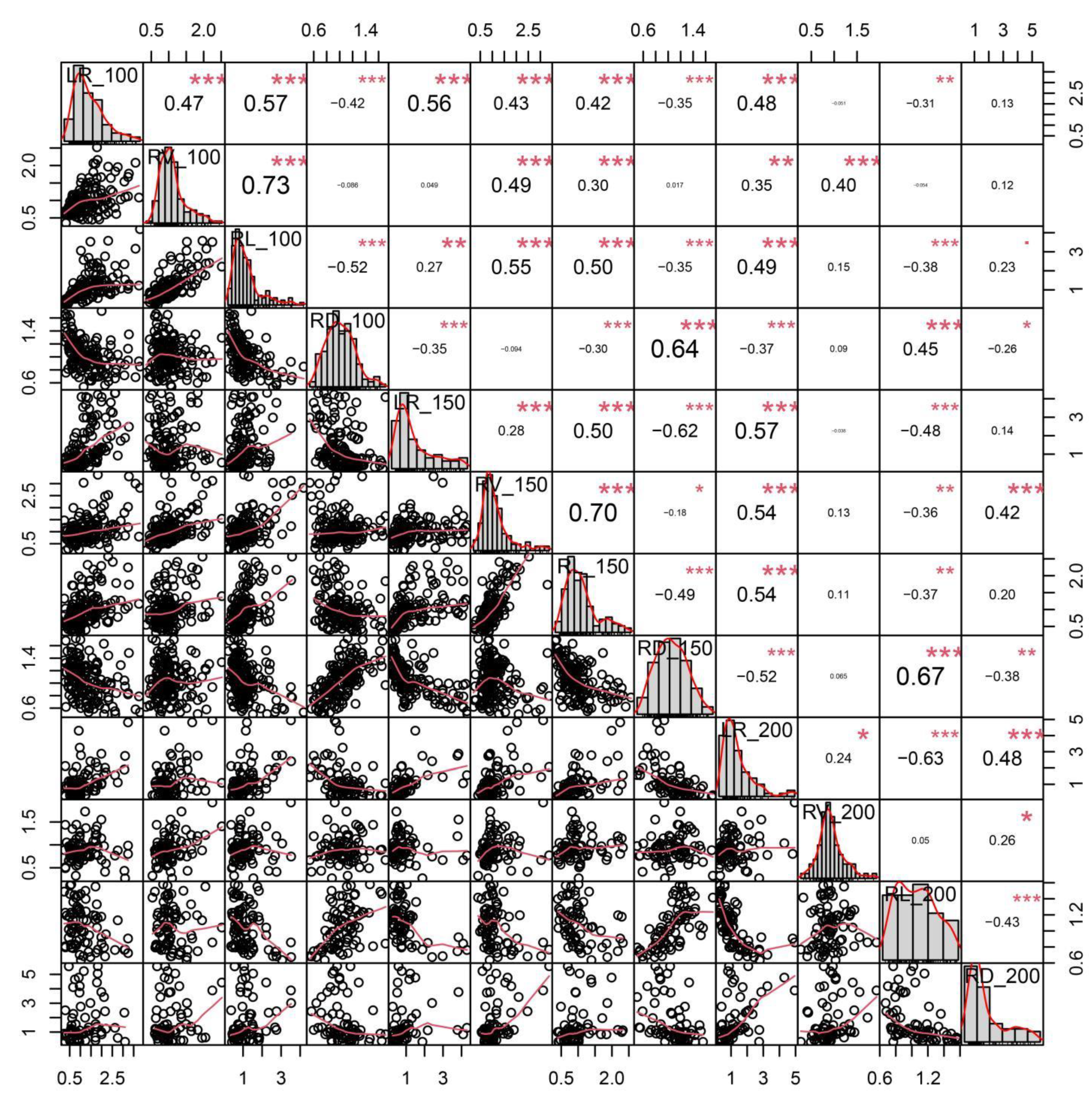
2.1. Analysis of Phenotypic Variations and Correlations among Salt-Related Root Traits
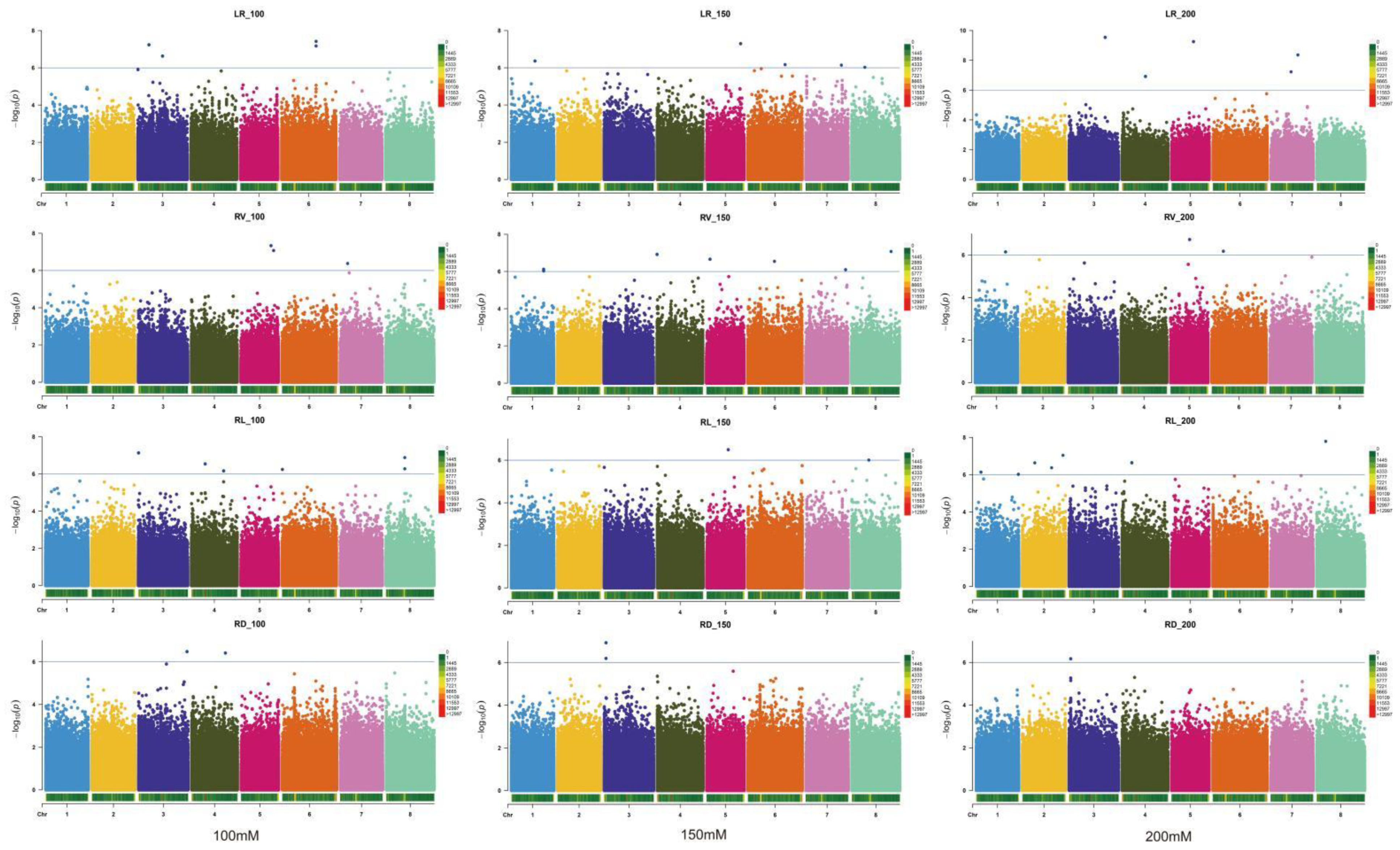
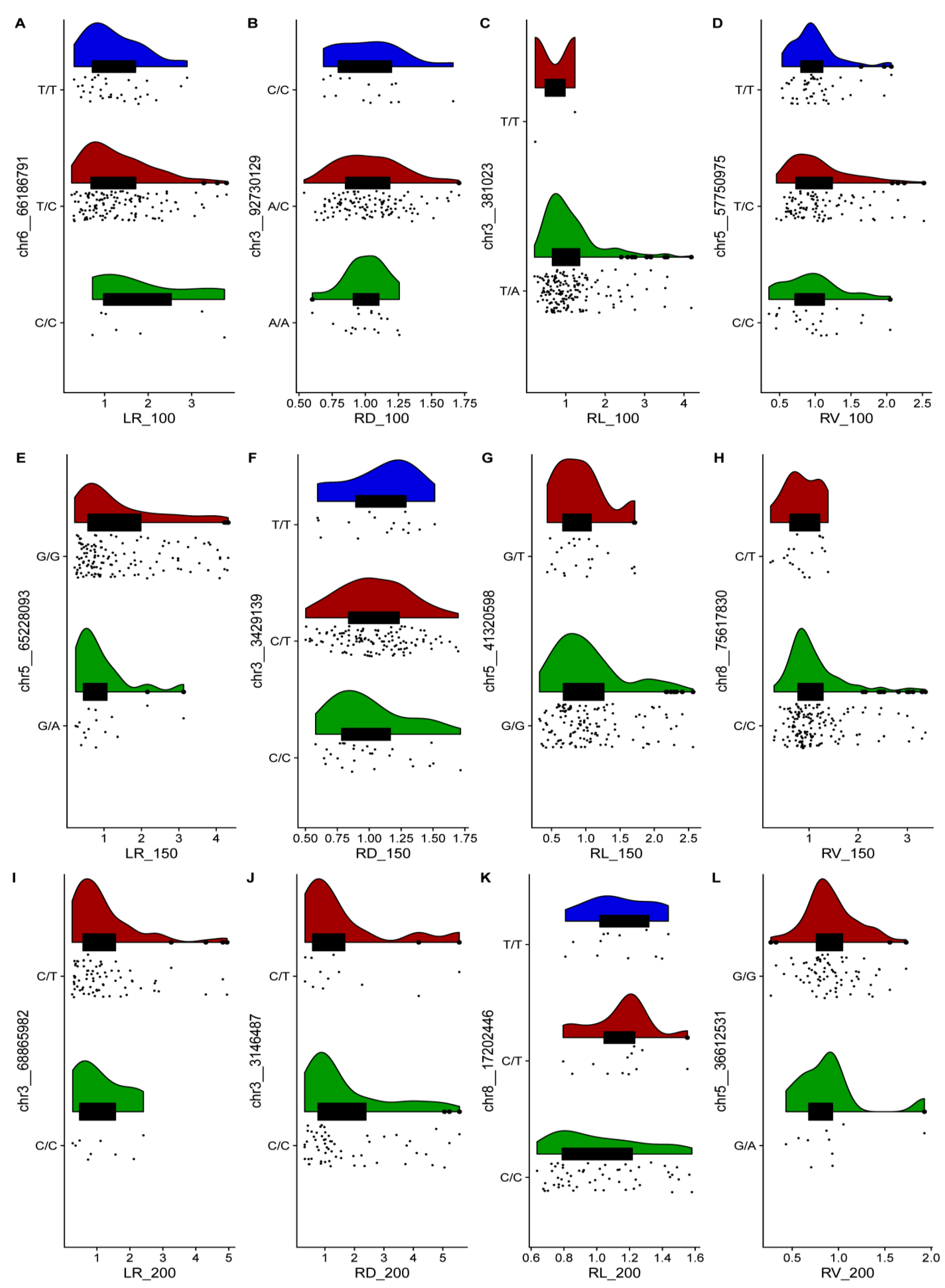
2.2. GWAS and Identification of Candidate Genes for 12 Relevant Phenotypic Traits
2.3. RNA-Seq Analysis
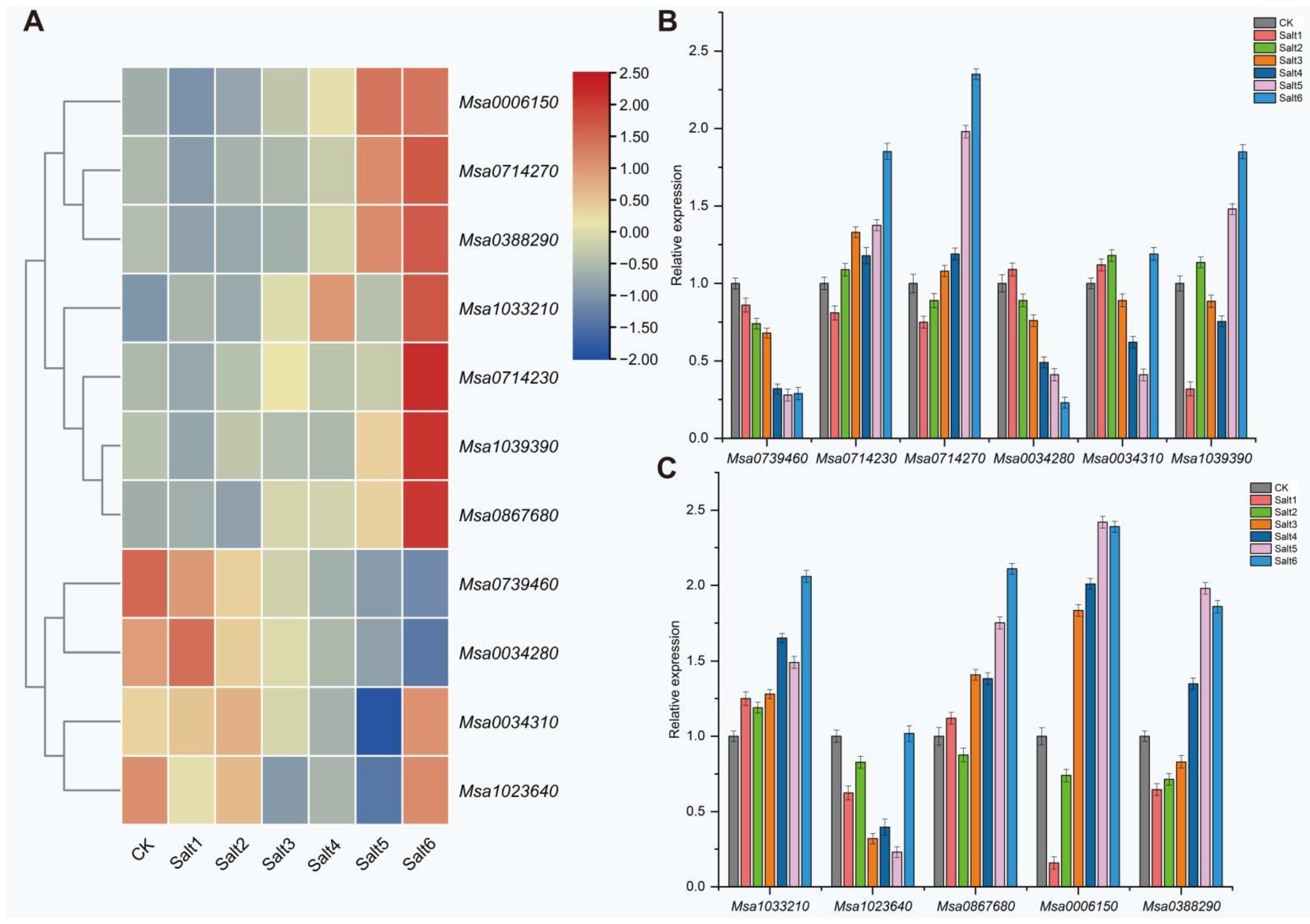
2.4. Candidate Gene Analysis Related to Salt Tolerance in Alfalfa
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Salt Stress Treatment and Phenotyping
4.3. Phenotypic Data Analysis
4.4. SNP Calling and Genome-Wide Association Studies
4.5. RNA-Seq and Transcriptomic Analysis
4.6. Candidate Gene Analysis and RT-qPCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sakadevan, K.; Nguyen, M.L. Extent, impact, and response to soil and water salinity in arid and semiarid regions. Adv. Agron. 2010, 109, 55–74. [Google Scholar]
- Tang, H.L.; Niu, L.; Wei, J.; Chen, X.Y.; Chen, Y.L. Phosphorus Limitation Improved Salt Tolerance in Maize through Tissue Mass Density Increase, Osmolytes Accumulation, and Na+ Uptake Inhibition. Front. Plant Sci. 2019, 10, 856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchanda, G.; Garg, N. Salinity and its effects on the functional biology of legumes. Acta Physiol. Plant. 2008, 30, 595–618. [Google Scholar] [CrossRef]
- Shannon, M.C.; Grieve, C.M. Tolerance of vegetable crops to salinity. Sci. Hortic. Amst. 1999, 78, 5–38. [Google Scholar] [CrossRef]
- Foolad, M.; Hyman, J.; Lin, G. Relationships between cold-and salt-tolerance during seed germination in tomato, Analysis of response and correlated response to selection. Plant Breed. 1999, 118, 49–52. [Google Scholar] [CrossRef]
- Yohannes, G.; Kidane, L.; Abraha, B.; Beyene, T. Effect of Salt Stresses on Seed Germination and Early Seedling Growth of Camelina sativa L. Momona Ethiop. J. Sci. 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Zhu, J.; Ingram, P.A.; Benfey, P.N.; Elich, T. From lab to field, new approaches to phenotyping root system architecture. Curr. Opin. Plant Biol. 2011, 14, 310–317. [Google Scholar] [CrossRef]
- Ma, Y.; Qiu, C.W.; Fan, Y.; Huang, X.; Khan, W.; Wu, F.; Zhou, M.; Wang, Y.; Cao, F. Genome-wide association and transcriptome analysis reveals candidate genes for potassium transport under salinity stress in wheat. Environ. Exp. Bot. 2022, 202, 105034. [Google Scholar] [CrossRef]
- Uga, Y.; Sugimoto, K.; Ogawa, S.; Rane, J.; Ishitani, M.; Hara, N.; Kitomi, Y.; Inukai, Y.; Ono, K.; Kanno, N.; et al. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 2013, 45, 1097–1102. [Google Scholar] [CrossRef]
- Landi, P.; Albrecht, B.; Giuliani, M.M.; Sanguineti, M.C. Seedling characteristics in hydroponic culture and field performance of maize genotypes with different resistance to root lodging. Maydica 1998, 43, 111–116. [Google Scholar]
- Bruce, W.; Desbons, P.; Crasta, O.; Folkerts, O. Gene expression profiling of two related maize inbred lines with contrasting root-lodging traits. J. Exp. Bot. 2001, 52, 459–468. [Google Scholar]
- Guo, J.; Li, C.H.; Zhang, X.Q.; Li, Y.X.; Zhang, D.; Shi, Y.; Song, Y.; Li, Y.; Yang, D.; Wang, T. Transcriptome and GWAS analyses reveal candidate gene for seminal root length of maize seedlings under drought stress. Plant Sci. 2020, 292, 110380. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Du, H.; Chen, Z.; Lu, H.; Zhu, F.; Chen, H.; Meng, X.; Liu, Q.; Liu, P.; Zheng, L.; et al. The chromosome-level genome sequence of the autotetraploid alfalfa and resequencing of core germplasms provide genomic resources for alfalfa research. Mol. Plant 2013, 13, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Long, R.C.; Yang, Q.C.; Kang, J.M.; Chao, Y.; Wang, P.; Wu, M.; Sun, Y. Molecular cloning and characterization of a novel stress responsive gene in alfalfa. Biol. Plant. 2012, 56, 43–49. [Google Scholar] [CrossRef]
- Qiao, G.; Zhang, X.; Jiang, J.; Liu, M.; Han, X.; Yang, H.; Zhuo, R. Comparative proteomic analysis of responses to salt stress in Chinese willow (Salix matsudana Koidz). Plant Mol. Biol. Rep. 2014, 32, 814–827. [Google Scholar] [CrossRef]
- Qi, X.; Li, M.W.; Xie, M.; Liu, X.; Ni, M.; Shao, G.; Song, C.; Kay-Yuen Yim, A.; Tao, Y.; Wong, F.L.; et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat. Commun. 2014, 5, 4340. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Zhang, Y.; Li, J.; Zhang, P.; Chen, K.; Song, W.; Wang, X.; Yang, J.; Lu, X.; Lu, B.; et al. Molecular dissection of maize seedling salt tolerance using a genome-wide association analysis method. Plant Biotechnol. J. 2021, 19, 1937–1951. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Gou, Z.; Zhang, Y.; Wang, X.; Ren, H.; Wen, Z.; Kang, B.-K.; Li, Y.; Yu, L.; et al. Genome-wide association study of soybean seed germination under drought stress. Mol. Genet. Genom. 2020, 295, 661–673. [Google Scholar] [CrossRef]
- He, F.; Wei, C.X.; Zhang, Y.X.; Long, R.; Li, M.; Wang, Z.; Yang, Q.; Kang, J.; Chen, L. Genome-wide association analysis coupled with transcriptome analysis reveals candidate genes related to salt stress in alfalfa (Medicago sativa L.). Front. Plant Sci. 2022, 12, 826584. [Google Scholar] [CrossRef]
- Zhang, T.J.; Yu, L.X.; Zheng, P.; Li, Y.; Rivera, M.; Main, D.; Greene, S.L. Identification of loci associated with drought resistance traits in heterozygous autotetraploid alfalfa (Medicago sativa L.) using genome-wide association studies with genotyping by sequencing. PLoS ONE 2015, 10, e0138931. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Medina, C.A.; Boge, B.; Hu, J.; Fransen, S.; Norberg, S.; Yu, L.-X. Identification of genetic loci associated with forage quality in response to water deficit in autotetraploid alfalfa (Medicago sativa L.). BMC Plant Biol. 2020, 20, 303. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.; Zhang, H.; Ma, L.; Zhao, H.; Jones, C.S.; Chen, J.; Liu, G. A genome-wide association study approach to the identification of candidate genes underlying agronomic traits in alfalfa (Medicago sativa L.). Plant Biotechnol. J. 2020, 18, 611–613. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zeng, Y.; Yang, Y.; Huang, L.; Qiu, Q. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 2020, 11, 2494. [Google Scholar] [CrossRef] [PubMed]
- Long, R.C.; Zhang, F.; Zhang, Z.W.; Li, M.; Chen, L.; Wang, X.; Liu, W.; Zhang, T.; Yu, L.-X.; He, F.; et al. Genome assembly of alfalfa cultivar zhongmu-4 and identification of SNPs associated with agronomic traits. Genom. Proteom. Bioinform. 2022, 20, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Sahi, C.; Singh, A.; Kumar, K.; Blumwald, E.; Grover, A. Salt stress response in rice: Genetics, molecular biology, and comparative genomics. Funct. Integr. Genom. 2006, 6, 263–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, Z.; Li, C.; Xing, Y.; Luo, Y.; Wang, X.; Cai, H. Overexpression of MsRCI2D and MsRCI2E Enhances Salt Tolerance in Alfalfa (Medicago sativa L.) by Stabilizing Antioxidant Activity and Regulating Ion Homeostasis. Int. J. Mol. Sci. 2022, 23, 9810. [Google Scholar] [CrossRef]
- Li, J.; Ma, M.; Sun, Y.; Lu, P.; Shi, H.; Guo, Z.; Zhu, H. Comparative Physiological and Transcriptome Profiles Uncover Salt Tolerance Mechanisms in Alfalfa. Front. Plant Sci. 2022, 13, 931619. [Google Scholar] [CrossRef]
- Shahzadi, A.K.; Bano, H.; Ogbaga, C.C.; Ayyaz, A.; Parveen, R.; Zafar, Z.U.; Athar, H.-U.; Ashraf, M. Coordinated impact of ion exclusion, antioxidants and photosynthetic potential on salt tolerance of ridge gourd [Luffa acutangula (L.) Roxb.]. Plant Physiol. Biochem. 2021, 167, 517–528. [Google Scholar] [CrossRef]
- Pierik, R.; Testerink, C. The art of being fexible: How to escape from shade, salt, and drought. Plant Physiol. 2014, 166, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.T.; Giehl, R.F.; Meyer, R.C.; Altmann, T.; von Wirén, N. Natural variation of BSK3 tunes brassinosteroid signaling to regulate root foraging under low nitrogen. Nat. Commun. 2019, 10, 2378. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, M.; Göschl, C.; Dietzen, C.; Andersen, S.U.; Kopriva, S.; Busch, W. Identifcation of novel genes involved in phosphate accumulation in Lotus japonicus through Genome Wide Association mapping of root system architecture and anion content. PLoS Genet. 2019, 15, e1008126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deolu-Ajayi, A.O.; Meyer, A.J.; Haring, M.A.; Julkowska, M.M.; Testerink, C. Genetic loci associated with early root responses to salt stress. iScience 2019, 21, 458–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Zhang, J.H.; Li, W.Z.; Hu, W.; Duan, L.; Feng, Y.; Qiu, H.; Yue, B. Genome-wide association analysis of ten cold tolerance indices at the germination and seeding stages in maize. J. Integr. Plant Biol. 2013, 55, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Nagel, K.A.; Kastenholz, B.; Jahnke, S.; van Dusschoten, D.; Aach, T.; Mühlich, M.; Truhn, D.; Scharr, H.; Terjung, S.; Walter, A.; et al. Temperature responses of roots: Impact on growth, root system architecture and implications for phenotyping. Funct. Plant Biol. 2009, 36, 947–959. [Google Scholar] [CrossRef]
- Osthoff, A.; Baldauf, J.A.; Piepho, H.-P.; Hochholdinger, F. Transcriptomic reprogramming of barley seminal roots by combined water deficit and salt stress. BMC Genom. 2019, 20, 325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kong, X.; Xu, X.; Li, C.; Tian, H.; Ding, Z. Comparative transcriptome profiling of the maize primary, crown and seminal root in response to salinity stress. PLoS ONE 2015, 10, e0121222. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.X.; Liu, X.; Boge, W.; Liu, X.-P. Genome-wide association study identifies loci for salt tolerance during germination in autotetraploid alfalfa (Medicago sativa L.) using genotyping-by-sequencing. Front. Plant Sci. 2016, 7, 956. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.P.; Yu, L.X. Genome-wide association mapping of loci associated with plant growth and forage production under salt stress in alfalfa (Medicago sativa L.). Front. Plant Sci. 2017, 8, 853. [Google Scholar] [CrossRef]
- Tai, H.H.; Lu, X.; Opitz, N.; Marcon, C.; Paschold, A.; Lithio, A.; Nettleton, D.; Hochholdinger, F. Transcriptomic and anatomical complexity of primary, seminal, and crown roots highlight root type-specific functional diversity in maize (Zea mays L.). J. Exp. Bot. 2016, 67, 1123–1135. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, J.; Xu, Q.; Wang, D.; Di, H.; Huang, J.; Yang, X.; Wang, Z.; Zhang, L.; Dong, L.; et al. Identification of candidate tolerance genes to low-temperature during maize germination by GWAS and RNA-seq approaches. BMC Plant Biol. 2020, 20, 333. [Google Scholar] [CrossRef]
- Cheng, Q.M.; Bai, S.Q.; Ge, G.T.; Li, P.; Liu, L.; Zhang, C.; Jia, Y. Study on differentially expressed genes related to defoliation traits in two alfalfa varieties based on RNA-Seq. BMC Genom. 2018, 19, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshad, M.; Gruber, M.Y.; Hannoufa, A. Transcriptome analysis of microRNA156 overexpression alfalfa roots under drought stress. Sci. Rep. 2018, 8, 9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postnikova, O.A.; Shao, J.; Nemchinov, L.G. Analysis of the alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol. 2013, 54, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, H.; Song, C.; Zhu, J.K.; Shabala, S. Mechanisms of plant responses and adaptation to soil salinity. Innovation-Amsterdam 2020, 1, 100017. [Google Scholar] [CrossRef]
- Missihoun, T.D.; Willée, E.; Guegan, J.P.; Guegan, J.P.; Berardocco, S.; Shafiq, M.R.; Bouchereau, A.; Bartels, D. Overexpression of ALDH10A8 and ALDH10A9 genes provides insight into their role in glycine betaine synthesis and affects primary metabolism in Arabidopsis thaliana. Plant Cell Physiol. 2015, 56, 1798–1807. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; He, F.; Long, R.C.; Zhang, F.; Li, M.N.; Wang, Z.; Kang, J.M.; Yang, Q.C. A global alfalfa diversity panel reveals genomic selection signatures in Chinese varieties and genomic associations with root development. J. Integr. Plant Biol. 2021, 63, 1937–1951. [Google Scholar] [CrossRef]
- Fischer, R.; Maurer, R. Drought resistance in spring wheat cultivars. I. Grain yield responses. Aust. J. Agric. Res. 1978, 29, 897–912. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. GigaScience 2019, 8, giy154. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.M.; Deng, H.; Ma, W.X.; Qiang, Z.; Liu, Z.P. Genome-wide identification of the MADS-box transcription factor family in autotetraploid cultivated alfalfa (Medicago sativa L.) and expression analysis under abiotic stress. BMC Genom. 2021, 22, 603. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Median | Mean | Range | SD | Kurtosis | Skewness | CV |
|---|---|---|---|---|---|---|---|
| LR_100 | 1.12 | 1.31 | 0.24–3.80 | 0.78 | 0.77 | 1.06 | 0.59 |
| RV_100 | 0.96 | 1.03 | 0.35–2.52 | 0.40 | 1.45 | 1.21 | 0.39 |
| RL_100 | 0.94 | 1.14 | 0.21–4.19 | 0.71 | 1.12 | 1.74 | 0.63 |
| RD_100 | 1.00 | 1.03 | 0.54–1.71 | 0.24 | 0.02 | 0.44 | 0.43 |
| LR_150 | 0.91 | 1.37 | 0.21–4.32 | 1.12 | 0.33 | 1.20 | 0.82 |
| RV_150 | 0.94 | 1.11 | 0.22–3.37 | 0.59 | 1.08 | 1.65 | 0.53 |
| RL_150 | 0.92 | 1.02 | 0.32–2.57 | 0.50 | 0.70 | 1.09 | 0.49 |
| RD_150 | 1.02 | 1.04 | 0.50–1.72 | 0.28 | −0.60 | 0.20 | 0.37 |
| LR_200 | 0.91 | 1.21 | 0.25–4.95 | 0.99 | 4.23 | 1.93 | 0.81 |
| RV_200 | 0.88 | 0.91 | 0.27–1.93 | 0.29 | 1.44 | 0.76 | 0.32 |
| RL_200 | 1.04 | 1.06 | 0.64–1.58 | 0.26 | −0.95 | 0.30 | 0.44 |
| RD_200 | 1.11 | 1.72 | 0.31–5.57 | 1.48 | 0.51 | 1.30 | 0.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, F.; Yang, T.; Zhang, F.; Jiang, X.; Li, X.; Long, R.; Wang, X.; Gao, T.; Wang, C.; Yang, Q.; et al. Transcriptome and GWAS Analyses Reveal Candidate Gene for Root Traits of Alfalfa during Germination under Salt Stress. Int. J. Mol. Sci. 2023, 24, 6271. https://doi.org/10.3390/ijms24076271
He F, Yang T, Zhang F, Jiang X, Li X, Long R, Wang X, Gao T, Wang C, Yang Q, et al. Transcriptome and GWAS Analyses Reveal Candidate Gene for Root Traits of Alfalfa during Germination under Salt Stress. International Journal of Molecular Sciences. 2023; 24(7):6271. https://doi.org/10.3390/ijms24076271
Chicago/Turabian StyleHe, Fei, Tianhui Yang, Fan Zhang, Xueqian Jiang, Xianyang Li, Ruicai Long, Xue Wang, Ting Gao, Chuan Wang, Qingchuan Yang, and et al. 2023. "Transcriptome and GWAS Analyses Reveal Candidate Gene for Root Traits of Alfalfa during Germination under Salt Stress" International Journal of Molecular Sciences 24, no. 7: 6271. https://doi.org/10.3390/ijms24076271