
The Role of Rab Proteins in Mitophagy: Insights into Neurodegenerative Diseases


Abstract
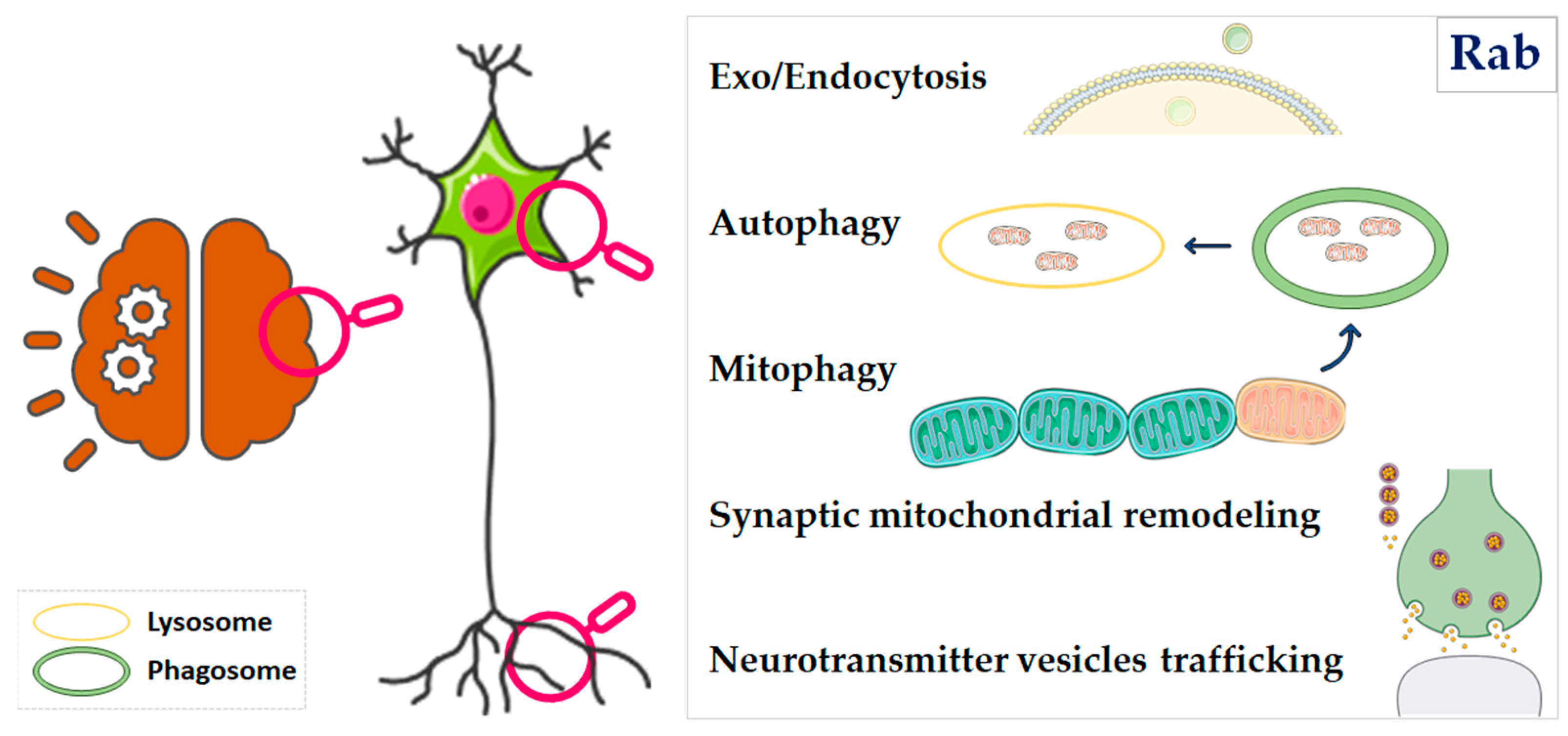
:1. Introduction
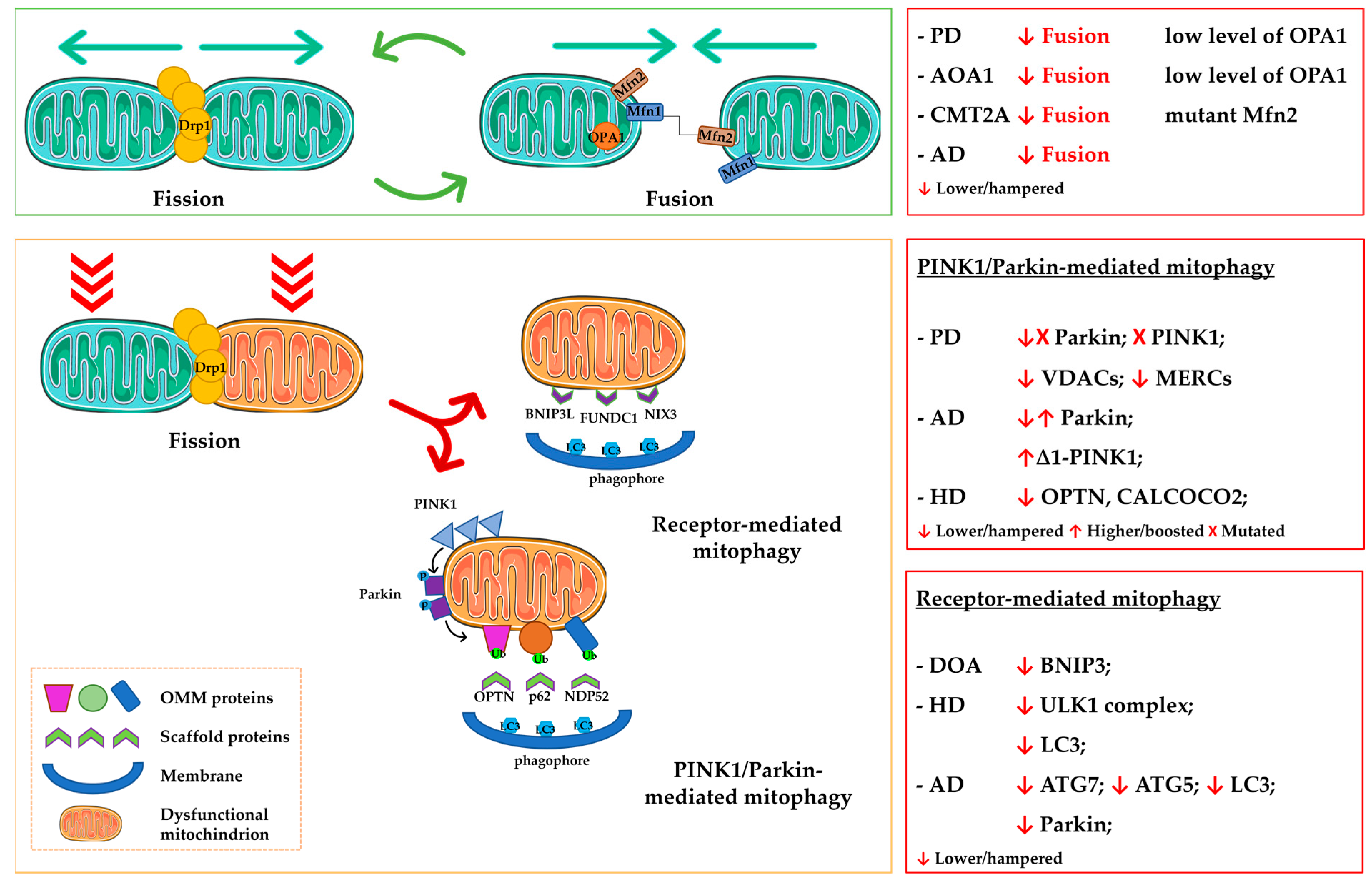
2. Mitochondrial Dynamics Altered in Neurodegeneration
2.1. Fusion and Fission
2.2. Mitophagy
3. Rab Proteins at the Crossroads of Mitochondrial Dysfunction and Neurodegeneration
3.1. The Role of Rab Proteins in Canonical PINK1/Parkin-Mediated Mitophagy
3.2. The Role of Rab Proteins in Non-Canonical Mitophagy
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Huang, T.; Bu, G.; Xu, H. Dysregulation of Protein Trafficking in Neurodegeneration. Mol. Neurodegener. 2014, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [Green Version]
- Blackstone, C.; Elwood, F.; Plun-Favreau, H.; Lewis, P.A. Vesicle Trafficking and Pathways to Neurodegeneration. Mol. Neurodegener. 2021, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.S.C.; DiGiovanni, L.; Kumar, R.; Carmichael, R.E.; Kim, P.K.; Schrader, M. Maintaining Social Contacts: The Physiological Relevance of Organelle Interactions. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118800. [Google Scholar] [CrossRef]
- Gordaliza-Alaguero, I.; Cantó, C.; Zorzano, A. Metabolic Implications of Organelle–Mitochondria Communication. EMBO Rep. 2019, 20, e47928. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, G.J.; Burns, M.E.; DeBello, W.M.; Hilfiker, S.; Morgan, J.R.; Schweizer, F.E.; Tokumaru, H.; Umayahara, K. Proteins Involved in Synaptic Vesicle Trafficking. J. Physiol. 1999, 520, 33–41. [Google Scholar] [CrossRef]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic Mitochondria in Synaptic Transmission and Organization of Vesicle Pools in Health and Disease. Front. Synaptic Neurosci. 2010, 2, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebanks, K.; Lewis, P.A.; Bandopadhyay, R. Vesicular Dysfunction and the Pathogenesis of Parkinson’s Disease: Clues From Genetic Studies. Front. Neurosci. 2020, 13, 1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.; Jeong, H.; Kwon, J.; Heo, H.Y.; Kwon, J.J.; Yun, H.J.; Kim, C.-H.; Han, B.S.; Tong, Y.; Shen, J.; et al. LRRK2 Regulates Synaptic Vesicle Endocytosis. Exp. Cell Res. 2008, 314, 2055–2065. [Google Scholar] [CrossRef]
- Liu, Z.; Bryant, N.; Kumaran, R.; Beilina, A.; Abeliovich, A.; Cookson, M.R.; West, A.B. LRRK2 Phosphorylates Membrane-Bound Rabs and Is Activated by GTP-Bound Rab7L1 to Promote Recruitment to the Trans-Golgi Network. Hum. Mol. Genet. 2018, 27, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264. [Google Scholar] [CrossRef]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin Ubiquitinates the Alpha-Synuclein-Interacting Protein, Synphilin-1: Implications for Lewy-Body Formation in Parkinson Disease. Nat. Med. 2001, 7, 1144–1150. [Google Scholar] [CrossRef]
- Kia, D.A.; Zhang, D.; Guelfi, S.; Manzoni, C.; Hubbard, L.; United Kingdom Brain Expression Consortium (UKBEC); International Parkinson’s Disease Genomics Consortium (IPDGC); Reynolds, R.H.; Botía, J.; Ryten, M.; et al. Integration of EQTL and Parkinson’s Disease GWAS Data Implicates 11 Disease Genes. bioRxiv 2019, 10, 627216. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, T.B. Focusing on Mitochondrial Form and Function. Nat. Cell Biol. 2018, 20, 735. [Google Scholar] [CrossRef] [Green Version]
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, T.B. The Functional Impact of Mitochondrial Structure Across Subcellular Scales. Front. Physiol. 2020, 11, 541040. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of Mitophagy in Cellular Homeostasis, Physiology and Pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated Mitophagy and Mitochondrial Organization in Optic Atrophy Due to OPA1 Mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Ramonet, D.; Perier, C.; Recasens, A.; Dehay, B.; Bové, J.; Costa, V.; Scorrano, L.; Vila, M. Optic Atrophy 1 Mediates Mitochondria Remodeling and Dopaminergic Neurodegeneration Linked to Complex I Deficiency. Cell Death Differ. 2013, 20, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Croteau, D.L.; Bohr, V.A.; Akbari, M. Diminished OPA1 Expression and Impaired Mitochondrial Morphology and Homeostasis in Aprataxin-Deficient Cells. Nucleic Acids Res. 2019, 47, 4086–4110. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Saubouny, R.; Joel, M.M.; Martens, K.; Martino, D.; de Koning, A.P.J.; Pfeffer, G.; Shutt, T.E. Characterization of a Novel Variant in the HR1 Domain of MFN2 in a Patient with Ataxia, Optic Atrophy and Sensorineural Hearing Loss. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.K.M.G.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Restoring Mitofusin Balance Prevents Axonal Degeneration in a Charcot-Marie-Tooth Type 2A Model. J. Clin. Investig. 2019, 129, 1756–1771. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Harland, M.; Torres, S.; Liu, J.; Wang, X. Neuronal Mitochondria Modulation of LPS-Induced Neuroinflammation. J. Neurosci. 2020, 40, 1756–1765. [Google Scholar] [CrossRef]
- Batista, A.F.; Rody, T.; Forny-Germano, L.; Cerdeiro, S.; Bellio, M.; Ferreira, S.T.; Munoz, D.P.; De Felice, F.G. Interleukin-1β Mediates Alterations in Mitochondrial Fusion/Fission Proteins and Memory Impairment Induced by Amyloid-β Oligomers. J. Neuroinflamm. 2021, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Fon, E.A. Principles of Mitochondrial Vesicle Transport. Curr. Opin. Physiol. 2018, 3, 25–33. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin Is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Zilocchi, M.; Colugnat, I.; Lualdi, M.; Meduri, M.; Marini, F.; Corasolla Carregari, V.; Moutaoufik, M.T.; Phanse, S.; Pieroni, L.; Babu, M.; et al. Exploring the Impact of PARK2 Mutations on the Total and Mitochondrial Proteome of Human Skin Fibroblasts. Front. Cell Dev. Biol. 2020, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-Mediated Mitophagy Is Compromised by PD-Associated Mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-Dependent Recruitment of Parkin to Mitochondria in Mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiesel, F.C.; Fričová, D.; Hayes, C.S.; Coban, M.A.; Hudec, R.; Bredenberg, J.M.; Broadway, B.J.; Markham, B.N.; Yan, T.; Boneski, P.K.; et al. Substitution of PINK1 Gly411 Modulates Substrate Receptivity and Turnover. Autophagy 2022, 1–22. [Google Scholar] [CrossRef]
- Tufi, R.; Clark, E.H.; Hoshikawa, T.; Tsagkaraki, C.; Stanley, J.; Takeda, K.; Staddon, J.M.; Briston, T. High-Content Phenotypic Screen to Identify Small Molecule Enhancers of Parkin-Dependent Ubiquitination and Mitophagy. SLAS Discov. 2023. [Google Scholar] [CrossRef]
- Clark, E.H.; Vázquez de la Torre, A.; Hoshikawa, T.; Briston, T. Targeting Mitophagy in Parkinson’s Disease. J. Biol. Chem. 2021, 296, 100209. [Google Scholar] [CrossRef]
- Bondi, H.; Zilocchi, M.; Mare, M.G.; D’Agostino, G.; Giovannardi, S.; Ambrosio, S.; Fasano, M.; Alberio, T. Dopamine Induces Mitochondrial Depolarization without Activating PINK1-Mediated Mitophagy. J. Neurochem. 2015, 136, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Alberio, T.; Mammucari, C.; D’Agostino, G.; Rizzuto, R.; Fasano, M. Altered Dopamine Homeostasis Differentially Affects Mitochondrial Voltage-Dependent Anion Channels Turnover. Biochim. Biophys. Acta 2014, 1842, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.J.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between Mitophagy and Apoptosis by Parkin via VDAC1 Ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef]
- He, Y.; Wang, W.; Yang, T.; Thomas, E.R.; Dai, R.; Li, X. The Potential Role of Voltage-Dependent Anion Channel in the Treatment of Parkinson’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 4665530. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-Mitochondria Contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s Disease: Molecular Defects and Therapeutic Approaches. Mol. Psychiatry 2023, 28, 202–216. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-Associated Tau Impairs Mitophagy by Inhibiting Parkin Translocation to Mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 Enhancement Is Able to Compensate Mitophagy Alterations Found in Sporadic Alzheimer’s Disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Fedorowicz, M.A.; de Vries-Schneider, R.L.A.; Rüb, C.; Becker, D.; Huang, Y.; Zhou, C.; Alessi Wolken, D.M.; Voos, W.; Liu, Y.; Przedborski, S. Cytosolic Cleaved PINK1 Represses Parkin Translocation to Mitochondria and Mitophagy. EMBO Rep. 2014, 15, 86–93. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and Mitophagy Biomarkers Are Reduced in Sera of Patients with Alzheimer’s Disease and Mild Cognitive Impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goiran, T.; Duplan, E.; Chami, M.; Bourgeois, A.; El Manaa, W.; Rouland, L.; Dunys, J.; Lauritzen, I.; You, H.; Stambolic, V.; et al. β-Amyloid Precursor Protein Intracellular Domain Controls Mitochondrial Function by Modulating Phosphatase and Tensin Homolog-Induced Kinase 1 Transcription in Cells and in Alzheimer Mice Models. Biol. Psychiatry 2018, 83, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhang, T.; Ge, X.; Chen, J.; Zhao, Y.; Fu, J. Parkin Overexpression Attenuates Aβ-Induced Mitochondrial Dysfunction in HEK293 Cells by Restoring Impaired Mitophagy. Life Sci. 2020, 244, 117322. [Google Scholar] [CrossRef]
- Randow, F.; Youle, R.J. Self and Nonself: How Autophagy Targets Mitochondria and Bacteria. Cell Host Microbe 2014, 15, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Kikuchi, R.; Kojima, W.; Hayashida, R.; Koyano, F.; Kawawaki, J.; Shoda, T.; Demizu, Y.; Naito, M.; Tanaka, K.; et al. Critical Role of Mitochondrial Ubiquitination and the OPTN–ATG9A Axis in Mitophagy. J. Cell Biol. 2020, 219, e201912144. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (Huntingtin) Impairs Mitophagy in a Cellular Model of Huntington Disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef]
- Terešak, P.; Lapao, A.; Subic, N.; Boya, P.; Elazar, Z.; Simonsen, A. Regulation of PRKN-Independent Mitophagy. Autophagy 2022, 18, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of Mitophagy Receptor BNIP3L/NIX Is Essential for Recruitment of Autophagic Machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia Induces the Expression of the Pro-Apoptotic Gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-Only Bnip3 Binds to the Dynamin Opa1 to Promote Mitochondrial Fragmentation and Apoptosis by Distinct Mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulis, M.F.; Millet, A.M.; Daloyau, M.; Miquel, M.-C.; Ronsin, B.; Wissinger, B.; Arnauné-Pelloquin, L.; Belenguer, P. OPA1 Haploinsufficiency Induces a BNIP3-Dependent Decrease in Mitophagy in Neurons: Relevance to Dominant Optic Atrophy. J. Neurochem. 2017, 140, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-W.; Yang, W.Y. Omegasome-Proximal PtdIns(4,5)P2 Couples F-Actin Mediated Mitoaggregate Disassembly with Autophagosome Formation during Mitophagy. Nat. Commun. 2019, 10, 969. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and Autophagy-Related Proteins in Cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Mercer, T.J.; Ohashi, Y.; Boeing, S.; Jefferies, H.B.J.; De Tito, S.; Flynn, H.; Tremel, S.; Zhang, W.; Wirth, M.; Frith, D.; et al. Phosphoproteomic Identification of ULK Substrates Reveals VPS15-dependent ULK/VPS34 Interplay in the Regulation of Autophagy. EMBO J. 2021, 40, e105985. [Google Scholar] [CrossRef]
- Zhou, C.; Ma, K.; Gao, R.; Mu, C.; Chen, L.; Liu, Q.; Luo, Q.; Feng, D.; Zhu, Y.; Chen, Q. Regulation of MATG9 Trafficking by Src- and ULK1-Mediated Phosphorylation in Basal and Starvation-Induced Autophagy. Cell Res. 2017, 27, 184–201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Guo, L.; Yang, Y. Mammalian ATG9s Drive the Autophagosome Formation by Binding to LC3. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The Autophagosome: Origins Unknown, Biogenesis Complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhou, J.; Wu, J.; Huang, J. ERβ Promotes Aβ Degradation via the Modulation of Autophagy. Cell Death Dis. 2019, 10, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara Ordóñez, A.J.; Fasiczka, R.; Naaldijk, Y.; Hilfiker, S. Rab GTPases in Parkinson’s Disease: A Primer. Essays Biochem. 2021, 65, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.P.; Goody, R.S. Molecular Control of Rab Activity by GEFs, GAPs and GDI. Small GTPases 2018, 9, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Bonet-Ponce, L.; Cookson, M.R. The Role of Rab GTPases in the Pathobiology of Parkinson’ Disease. Curr. Opin. Cell Biol. 2019, 59, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as Coordinators of Vesicle Traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Li, G. Rab GTPases, Membrane Trafficking and Diseases. Curr. Drug Targets 2011, 12, 1188–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Adamo, P.; Masetti, M.; Bianchi, V.; Morè, L.; Mignogna, M.L.; Giannandrea, M.; Gatti, S. RAB GTPases and RAB-Interacting Proteins and Their Role in the Control of Cognitive Functions. Neurosci. Biobehav. Rev. 2014, 46, 302–314. [Google Scholar] [CrossRef]
- Mignogna, M.L.; D’Adamo, P. Critical Importance of RAB Proteins for Synaptic Function. Small GTPases 2018, 9, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Giannandrea, M.; Bianchi, V.; Mignogna, M.L.; Sirri, A.; Carrabino, S.; D’Elia, E.; Vecellio, M.; Russo, S.; Cogliati, F.; Larizza, L.; et al. Mutations in the Small GTPase Gene RAB39B Are Responsible for X-Linked Mental Retardation Associated with Autism, Epilepsy, and Macrocephaly. Am. J. Hum. Genet. 2010, 86, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.R.; Sim, J.C.H.; McLean, C.; Giannandrea, M.; Galea, C.A.; Riseley, J.R.; Stephenson, S.E.M.; Fitzpatrick, E.; Haas, S.A.; Pope, K.; et al. Mutations in RAB39B Cause X-Linked Intellectual Disability and Early-Onset Parkinson Disease with α-Synuclein Pathology. Am. J. Hum. Genet. 2014, 95, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, M.; Zheng, N.; Wang, Z.; Gao, Y.; Luo, X.; Chen, Z.; Fu, X.; Wang, Y.; Wang, T.; Liu, M.; et al. RAB39B Deficiency Impairs Learning and Memory Partially Through Compromising Autophagy. Front. Cell Dev. Biol. 2020, 8, 598622. [Google Scholar] [CrossRef] [PubMed]
- Purlyte, E.; Dhekne, H.S.; Sarhan, A.R.; Gomez, R.; Lis, P.; Wightman, M.; Martinez, T.N.; Tonelli, F.; Pfeffer, S.R.; Alessi, D.R. Rab29 Activation of the Parkinson’s Disease-Associated LRRK2 Kinase. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingwell, K. LRRK2-Targeted Parkinson Disease Drug Advances into Phase III. Nat. Rev. Drug Discov. 2022, 22, 3–5. [Google Scholar] [CrossRef]
- Connor-Robson, N.; Booth, H.; Martin, J.G.; Gao, B.; Li, K.; Doig, N.; Vowles, J.; Browne, C.; Klinger, L.; Juhasz, P.; et al. An Integrated Transcriptomics and Proteomics Analysis Reveals Functional Endocytic Dysregulation Caused by Mutations in LRRK2. Neurobiol. Dis. 2019, 127, 512–526. [Google Scholar] [CrossRef]
- Zeigerer, A.; Gilleron, J.; Bogorad, R.L.; Marsico, G.; Nonaka, H.; Seifert, S.; Epstein-Barash, H.; Kuchimanchi, S.; Peng, C.G.; Ruda, V.M.; et al. Rab5 Is Necessary for the Biogenesis of the Endolysosomal System in Vivo. Nature 2012, 485, 465–470. [Google Scholar] [CrossRef]
- Sung, J.Y.; Kim, J.; Paik, S.R.; Park, J.H.; Ahn, Y.S.; Chung, K.C. Induction of Neuronal Cell Death by Rab5A-Dependent Endocytosis of α-Synuclein. J. Biol. Chem. 2001, 276, 27441–27448. [Google Scholar] [CrossRef] [Green Version]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 Induces Clearance of α-Synuclein Aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Yang, W.; Florio, J.B.; Rockenstein, E.; Spencer, B.; Orain, X.M.; Dong, S.X.; Li, H.; Chen, X.; Sung, K.; et al. Synuclein Impairs Trafficking and Signaling of BDNF in a Mouse Model of Parkinson’s Disease. Sci. Rep. 2017, 7, 3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breda, C.; Nugent, M.L.; Estranero, J.G.; Kyriacou, C.P.; Outeiro, T.F.; Steinert, J.R.; Giorgini, F. Rab11 Modulates α-Synuclein-Mediated Defects in Synaptic Transmission and Behaviour. Hum. Mol. Genet. 2015, 24, 1077–1091. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs Govern Autophagosome Biogenesis during Mitophagy. Elife 2014, 3, e01612. [Google Scholar] [CrossRef]
- Yamano, K.; Wang, C.; Sarraf, S.A.; Münch, C.; Kikuchi, R.; Noda, N.N.; Hizukuri, Y.; Kanemaki, M.T.; Harper, W.; Tanaka, K.; et al. Endosomal Rab Cycles Regulate Parkin-Mediated Mitophagy. Elife 2018, 7, e31326. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.-R.; Li, T.; Coyaud, E.; Laurent, E.M.N.; St-Germain, J.; Zhou, Y.; Kim, P.K.; Raught, B.; Brumell, J.H. C5orf51 Is a Component of the MON1-CCZ1 Complex and Controls RAB7A Localization and Stability during Mitophagy. Autophagy 2022, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Kabata, Y.; Morihara, T.; Ohara, T.; Ninomiya, T.; Takahashi, A.; Akatsu, H.; Hashizume, Y.; Hayashi, N.; Shigemizu, D.; Boroevich, K.A.; et al. Integrated Analysis of Human Genetic Association Study and Mouse Transcriptome Suggests LBH and SHF Genes as Novel Susceptible Genes for Amyloid-β Accumulation in Alzheimer’s Disease. Hum. Genet. 2018, 137, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Orgaz, A.; Kvainickas, A.; Nägele, H.; Denner, J.; Eimer, S.; Dengjel, J.; Steinberg, F. Control of RAB7 Activity and Localization through the Retromer-TBC1D5 Complex Enables RAB7-Dependent Mitophagy. EMBO J. 2018, 37, 235–254. [Google Scholar] [CrossRef]
- Heo, J.-M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A Phosphorylation by TBK1 Promotes Mitophagy via the PINK-PARKIN Pathway. Sci. Adv. 2018, 4, eaav0443. [Google Scholar] [CrossRef] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 Mitigate STING-Induced Inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Kim, S.; Wong, Y.C.; Gao, F.; Krainc, D. Dysregulation of Mitochondria-Lysosome Contacts by GBA1 Dysfunction in Dopaminergic Neuronal Models of Parkinson’s Disease. Nat. Commun. 2021, 12, 1807. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the Small GTP-Ase Late Endosomal Protein RAB7 Cause Charcot-Marie-Tooth Type 2B Neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioni, J.-M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as MRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-C.; Kondapalli, C.; Lehneck, R.; Procter, J.B.; Dill, B.D.; Woodroof, H.I.; Gourlay, R.; Peggie, M.; Macartney, T.J.; Corti, O.; et al. Phosphoproteomic Screening Identifies Rab GTPases as Novel Downstream Targets of PINK1. EMBO J. 2015, 34, 2840–2861. [Google Scholar] [CrossRef]
- Vieweg, S.; Mulholland, K.; Bräuning, B.; Kachariya, N.; Lai, Y.-C.; Toth, R.; Singh, P.K.; Volpi, I.; Sattler, M.; Groll, M.; et al. PINK1-Dependent Phosphorylation of Serine111 within the SF3 Motif of Rab GTPases Impairs Effector Interactions and LRRK2-Mediated Phosphorylation at Threonine72. Biochem. J. 2020, 477, 1651–1668. [Google Scholar] [CrossRef] [Green Version]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. LRRK2 Mutations Impair Depolarization-Induced Mitophagy through Inhibition of Mitochondrial Accumulation of RAB10. Autophagy 2020, 16, 203–222. [Google Scholar] [CrossRef]
- Yan, T.; Wang, L.; Gao, J.; Siedlak, S.L.; Huntley, M.L.; Termsarasab, P.; Perry, G.; Chen, S.G.; Wang, X. Rab10 Phosphorylation Is a Prominent Pathological Feature in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 157–165. [Google Scholar] [CrossRef]
- Minowa-Nozawa, A.; Nozawa, T.; Okamoto-Furuta, K.; Kohda, H.; Nakagawa, I. Rab35 GTPase Recruits NDP52 to Autophagy Targets. EMBO J. 2017, 36, 2790–2807. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Najor, R.H.; Cortez, M.Q.; Shires, S.E.; Leon, L.J.; Gonzalez, E.R.; Boassa, D.; Phan, S.; Thor, A.; Jimenez, R.E.; et al. A Rab5 Endosomal Pathway Mediates Parkin-Dependent Mitochondrial Clearance. Nat. Commun. 2017, 8, 14050. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Shires, S.E.; Leon, L.J.; Cortez, M.Q.; Gustafsson, Å.B. Isolation of Rab5-Positive Endosomes Reveals a New Mitochondrial Degradation Pathway Utilized by BNIP3 and Parkin. Small GTPases 2020, 11, 69–76. [Google Scholar] [CrossRef]
- Hsu, F.; Spannl, S.; Ferguson, C.; Hyman, A.A.; Parton, R.G.; Zerial, M. Rab5 and Alsin Regulate Stress-Activated Cytoprotective Signaling on Mitochondria. eLife 2018, 7, e32282. [Google Scholar] [CrossRef]
- Wu, K.K.L.; Long, K.; Lin, H.; Siu, P.M.F.; Hoo, R.L.C.; Ye, D.; Xu, A.; Cheng, K.K.Y. The APPL1-Rab5 Axis Restricts NLRP3 Inflammasome Activation through Early Endosomal-Dependent Mitophagy in Macrophages. Nat. Commun. 2021, 12, 6637. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, Y.; Yamashita, S.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy Is Primarily Due to Alternative Autophagy and Requires the MAPK1 and MAPK14 Signaling Pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, A.; Longhena, F.; Spillantini, M.G. The Role of Rab Proteins in Parkinson’s Disease Synaptopathy. Biomedicines 2022, 10, 1941. [Google Scholar] [CrossRef]
- Hall, C.N.; Klein-Flügge, M.C.; Howarth, C.; Attwell, D. Oxidative Phosphorylation, Not Glycolysis, Powers Presynaptic and Postsynaptic Mechanisms Underlying Brain Information Processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef] [PubMed]
- Monti, C.; Bondi, H.; Urbani, A.; Fasano, M.; Alberio, T. Systems Biology Analysis of the Proteomic Alterations Induced by MPP(+), a Parkinson’s Disease-Related Mitochondrial Toxin. Front. Cell. Neurosci. 2015, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataura, T.; Sedlackova, L.; Otten, E.G.; Kumari, R.; Shapira, D.; Scialo, F.; Stefanatos, R.; Ishikawa, K.; Kelly, G.; Seranova, E.; et al. Autophagy Promotes Cell Survival by Maintaining NAD Levels. Dev. Cell 2022, 57, 2584–2598.e11. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.L.; Koss, D.J.; Outeiro, T.F.; Giorgini, F. Therapeutic Targeting of Rab GTPases: Relevance for Alzheimer’s Disease. Biomedicines 2022, 10, 1141. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Rab Name | Pathways (Interactors) | References |
|---|---|---|
| Rab5A | Parkin-dependent micromitophagy via ESCRT machinery (BNIP3) | [109,110] |
| PINK1/Parkin-mediated mitophagy (RABGEF1/RABEX-5) | [94,95] | |
| Early endosome-mediated mitophagy (Rab5 and APPL1) | [112] | |
| Rab5, along with its effectors GEFs Alsin, implicated in ALS, and Rabenosyn-5, translocate to mitochondria upon stress to patch up damage, preceding mitophagy | [111] | |
| Rab7 | PINK1/Parkin-mediated mitophagy (MON1/CCZ1, TBC1D15, and TBC1D17) | [94,95] |
| PINK1/Parkin-mediated mitophagy via TBK1 phosphorylation of Rab7a | [99] | |
| C5orf51 mediates Rab7 localization to mitochondria during mitophagy | [96] | |
| Rab7 activity and localization to mitochondria regulated by TBC1D5 and the retromer complex | [98] | |
| Prolonged mitochondria-lysosome contacts in GBA1-mutant PD neurons due to defective Rab7-GTP hydrolysis (TBC1D15) | [101] | |
| Rab9 | Alternative mitophagy requires Rab9, MAPK1, and MAPK14 | [114] |
| MDV-mediated damaged cargo removal | [113] | |
| Rab10 | Rab10-mediated OPTN accumulation on mitochondria impaired due to LRRK2 mutations | [106] |
| Rab35 | TBK1-mediated targeting of NDP52 to damaged mitochondria | [108] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafique, A.; Brughera, M.; Lualdi, M.; Alberio, T. The Role of Rab Proteins in Mitophagy: Insights into Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 6268. https://doi.org/10.3390/ijms24076268
Shafique A, Brughera M, Lualdi M, Alberio T. The Role of Rab Proteins in Mitophagy: Insights into Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(7):6268. https://doi.org/10.3390/ijms24076268
Chicago/Turabian StyleShafique, Adeena, Martina Brughera, Marta Lualdi, and Tiziana Alberio. 2023. "The Role of Rab Proteins in Mitophagy: Insights into Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 7: 6268. https://doi.org/10.3390/ijms24076268