
Current Bioinformatics Tools to Optimize CRISPR/Cas9 Experiments to Reduce Off-Target Effects


Abstract
:1. Introduction
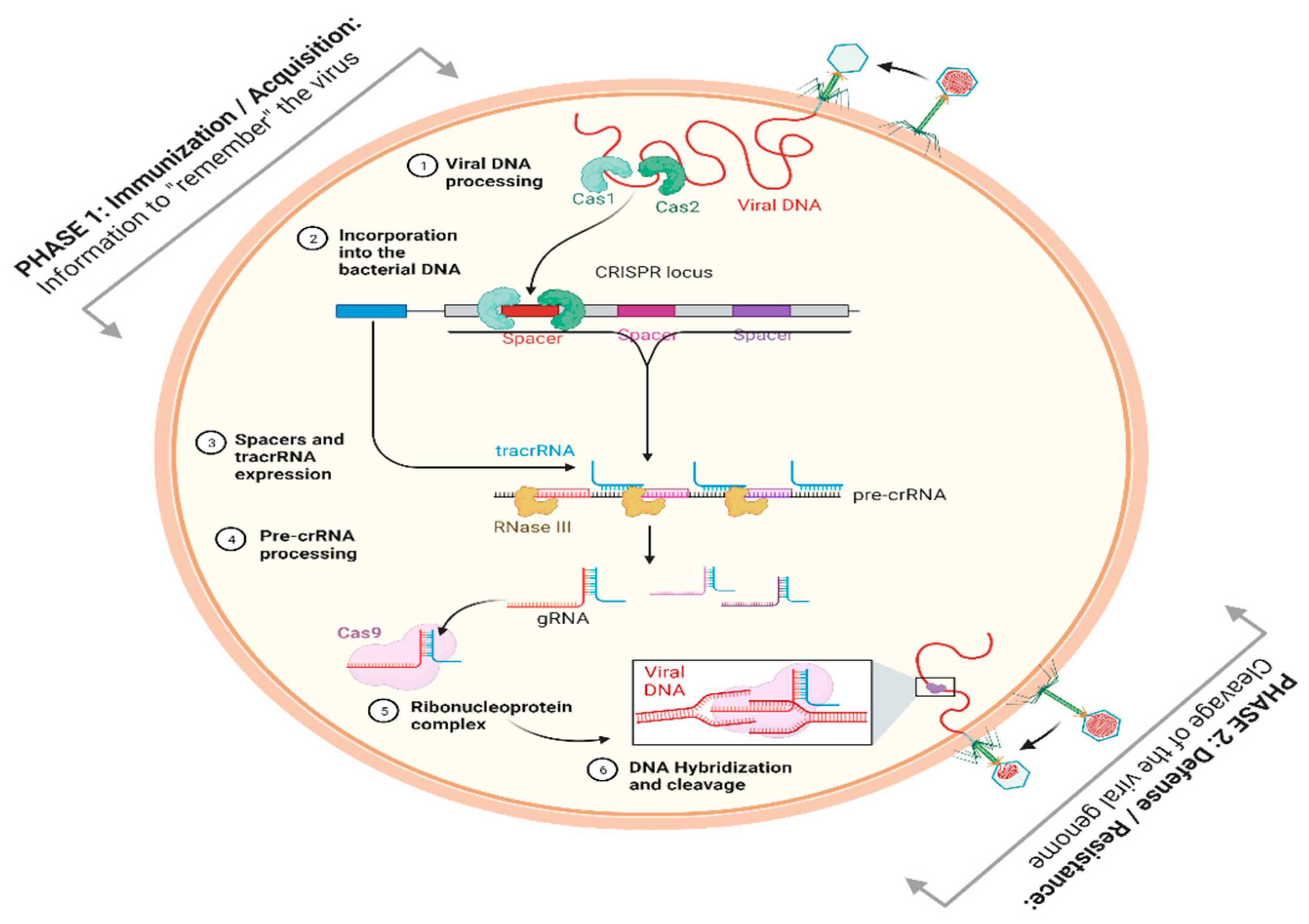
2. Role of Bioinformatics in the Development of CRISPR-Cas Technology
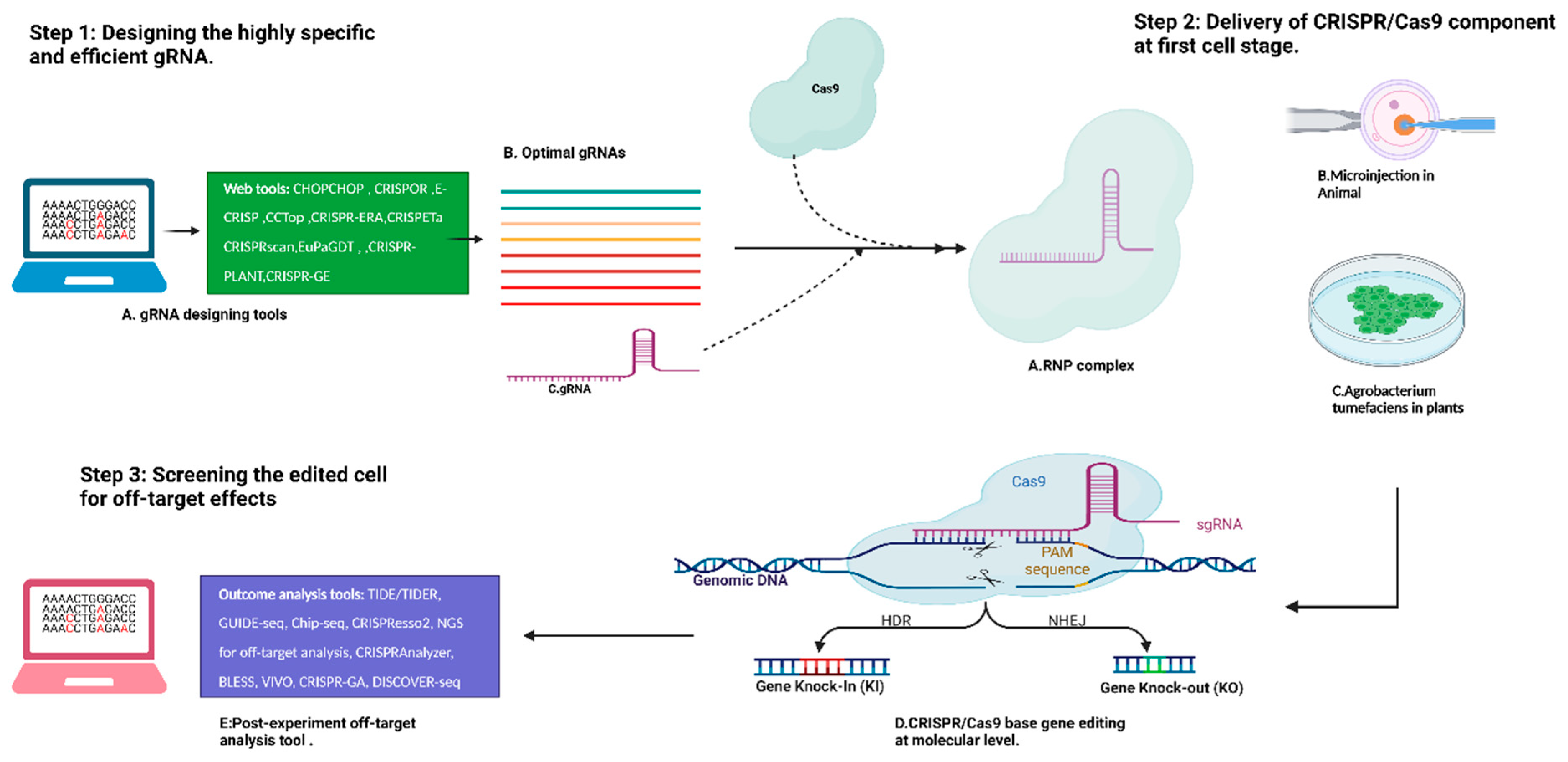
3. Optimized Workflow of CRISPR/Cas-System-Based Editing
4. Off-Target Detection Methods: Biased and Unbiased
Latest Biased and Unbiased Off-Target Detection Methods

{kind=link}
{kind=link}
| Tools | Description Features | Cell Line | Study | Limitations | Ref. |
|---|---|---|---|---|---|
| DeepCRISPR | Deep learning tool to predict off/on-target hits together with DNA methylation factors | Human and mouse cell lines | In vitro/ in vivo | Not suitable for base editors and prime editors | [26] |
| MOFF | The latest multi-layer regression-based model to predict off-target effects by incorporating the GMT and new epigenetic factors along with other factors, such as sequence features, structure features, and epigenetic features | Human and mouse cell lines | In vitro/ in vivo | Specificity | [27] |
| PEM-Seq | Latest generated off-target detection method, which is highly sensitive in detecting genomic translocations in edited cells | Human and mouse lines | In vivo | Not suitable for base editors and prime editors | [28] |
| GUIDE-Tag | Latest in vivo developed method to detect off-target effects where editing efficiencies are ≥0.2%. | Mouse and human cell lines | In vivo | Cannot provide specificity information | [29] |
| PEAC-Seq | Unbiased method of off-target effect identification in the prime-edited cells. | Mouse and human cell lines | In vivo | Sensitivity | [32] |
| TAPE-Seq | In vivo method to detect both on- and off-target events generated by prime editors | Human cell lines | In vivo | Sensitivity | [33] |
5. Efficient Guide RNA Design Tools
Properties to Consider for the Optimization of gRNA Design and Synthesis
6. Bioinformatics Tools for Repair Outcome Predictions
7. Bioinformatics for Post-CRISPR-Experiment Off-Target Analysis
8. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, H.; Farahani, N.; Hosseingholi, E.Z.; Sathyapalan, T.; Hossein Sahebkar, A. Harnessing CRISPR/Cas9 technology in cardiovascular disease. Trends Cardiovasc. Med. 2020, 30, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Alkhodairy, H.F.; Ashraf, I.; Khalil, A.B. CRISPR/Cas System Toward the Development of Next-Generation Recombinant Vaccines: Current Scenario and Future Prospects. Arab. J. Sci. Eng. 2022, 48, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Albadri, S.; Del Bene, F.; Revenu, C. Genome editing using CRISPR/Cas9-based knock-in approaches in zebrafish. Methods 2017, 121, 77–85. [Google Scholar] [CrossRef]
- Wang, B.; Li, K.; Wang, A.; Reiser, M.; Saunders, T.; Lockey, R.F.; Wang, J.-W. Highly efficient CRISPR/HDR-mediated knock-in for mouse embryonic stem cells and zygotes. Biotechniques 2015, 59, 201–208. [Google Scholar] [CrossRef]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest developed strategies to minimize the off-target effects in CRISPR-Cas-mediated genome editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef]
- Prykhozhij, S.V.; Rajan, V.; Berman, J.N. A guide to computational tools and design strategies for genome editing experiments in zebrafish using CRISPR/Cas9. Zebrafish 2016, 13, 70–73. [Google Scholar] [CrossRef]
- Ghorbani, A.; Hadifar, S.; Salari, R.; Izadpanah, K.; Burmistrz, M.; Afsharifar, A.; Eskandari, M.H.; Niazi, A.; Denes, C.E.; Neely, G.G. A short overview of CRISPR-Cas technology and its application in viral disease control. Transgenic Res. 2021, 30, 221–238. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Mojica, F.J.; Díez-Villaseñor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Alkhnbashi, O.S.; Meier, T.; Mitrofanov, A.; Backofen, R.; Voß, B. CRISPR-Cas bioinformatics. Methods 2020, 172, 3–11. [Google Scholar] [CrossRef]
- Luthra, R.; Kaur, S.; Bhandari, K. Applications of CRISPR as a potential therapeutic. Life Sci. 2021, 284, 119908. [Google Scholar] [CrossRef]
- Syombua, E.D.; Tripathi, J.N.; Obiero, G.O.; Nguu, E.K.; Yang, B.; Wang, K.; Tripathi, L. Potential applications of the CRISPR/Cas technology for genetic improvement of yam (Dioscorea spp.). Food Energy Secur. 2022, 11, e330. [Google Scholar] [CrossRef]
- Thomas, M.; Parry-Smith, D.; Iyer, V. Best practice for CRISPR design using current tools and resources. Methods 2019, 164, 3–17. [Google Scholar] [CrossRef]
- Yip, B.H. Recent advances in CRISPR/Cas9 delivery strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Rutz, S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, T.S.; Baudrier, L.; Billon, P.; Ciccia, A. CRISPR-based genome editing through the lens of DNA repair. Mol. Cell 2022, 82, 348–388. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Fu, R.; He, W.; Dou, J.; Villarreal, O.D.; Bedford, E.; Wang, H.; Hou, C.; Zhang, L.; Wang, Y.; Ma, D. Systematic decomposition of sequence determinants governing CRISPR/Cas9 specificity. Nat. Commun. 2022, 13, 474. [Google Scholar] [CrossRef]
- Liu, Y.; Yin, J.; Gan, T.; Liu, M.; Xin, C.; Zhang, W.; Hu, J. PEM-seq comprehensively quantifies DNA repair outcomes during gene-editing and DSB repair. STAR Protoc. 2022, 3, 101088. [Google Scholar] [CrossRef]
- Liang, S.-Q.; Liu, P.; Smith, J.L.; Mintzer, E.; Maitland, S.; Dong, X.; Yang, Q.; Lee, J.; Haynes, C.M.; Zhu, L.J. Genome-wide detection of CRISPR editing in vivo using GUIDE-tag. Nat. Commun. 2022, 13, 437. [Google Scholar] [CrossRef]
- Xu, C.-F.; Chen, G.-J.; Luo, Y.-L.; Zhang, Y.; Zhao, G.; Lu, Z.-D.; Czarna, A.; Gu, Z.; Wang, J. Rational designs of in vivo CRISPR-Cas delivery systems. Adv. Drug Deliv. Rev. 2021, 168, 3–29. [Google Scholar] [CrossRef]
- Zuo, E.; Sun, Y.; Wei, W.; Yuan, T.; Ying, W.; Sun, H.; Yuan, L.; Steinmetz, L.M.; Li, Y.; Yang, H. GOTI, a method to identify genome-wide off-target effects of genome editing in mouse embryos. Nat. Protoc. 2020, 15, 3009–3029. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lu, Z.; Li, J.; Wang, Y.; Wu, P.; Li, Y.; Zhou, Y.; Li, B.; Zhang, H.; Liu, Y. PEAC-seq adopts Prime Editor to detect CRISPR off-target and DNA translocation. Nat. Commun. 2022, 13, 7545. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Kim, M.; Bae, S.; Jo, A.; Kim, Y.; Lee, J.K. TAPE-seq is a cell-based method for predicting genome-wide off-target effects of prime editor. Nat. Commun. 2022, 13, 7975. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Yang, Z.; Xu, J.; Sun, J.; Mao, D.; Hu, Y.; Yang, S.-J.; Qiao, H.-H.; Wang, X.; Hu, Q. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep. 2014, 9, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017, 13, e1005807. [Google Scholar] [CrossRef]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef]
- Perez, A.R.; Pritykin, Y.; Vidigal, J.A.; Chhangawala, S.; Zamparo, L.; Leslie, C.S.; Ventura, A. GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 2017, 35, 347–349. [Google Scholar] [CrossRef]
- Ma, J.; Köster, J.; Qin, Q.; Hu, S.; Li, W.; Chen, C.; Cao, Q.; Wang, J.; Mei, S.; Liu, Q. CRISPR-DO for genome-wide CRISPR design and optimization. Bioinformatics 2016, 32, 3336–3338. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef]
- Peng, D.; Tarleton, R. EuPaGDT: A web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb. Genom. 2015, 1, e000033. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: Characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015, 16, 218. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Lu, L.; Liu, H.-Y.; Li, S.; Xing, F.; Chen, L.-L. CRISPR-P: A web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol. Plant 2014, 7, 1494–1496. [Google Scholar] [CrossRef] [PubMed]
- Varshney, G.K.; Pei, W.; LaFave, M.C.; Idol, J.; Xu, L.; Gallardo, V.; Carrington, B.; Bishop, K.; Jones, M.; Li, M. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015, 25, 1030–1042. [Google Scholar] [CrossRef]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef]
- Rastogi, A.; Murik, O.; Bowler, C.; Tirichine, L. PhytoCRISP-Ex: A web-based and stand-alone application to find specific target sequences for CRISPR/CAS editing. BMC Bioinform. 2016, 17, 261. [Google Scholar] [CrossRef]
- Siegner, S.M.; Karasu, M.E.; Schröder, M.S.; Kontarakis, Z.; Corn, J.E. PnB Designer: A web application to design prime and base editor guide RNAs for animals and plants. BMC Bioinform. 2021, 22, 101. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Adikaram, P.; Pandey, M.; Genis, A.; Simonds, W.F. Optimization of genome editing through CRISPR-Cas9 engineering. Bioengineered 2016, 7, 166–174. [Google Scholar] [CrossRef]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.P.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2018, 2, 38–47. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Zhang, T. Computational approaches for effective CRISPR guide RNA design and evaluation. Comput. Struct. Biotechnol. J. 2020, 18, 35–44. [Google Scholar] [CrossRef]
- Ma, J.-B.; Yuan, Y.-R.; Meister, G.; Pei, Y.; Tuschl, T.; Patel, D.J. Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature 2005, 434, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Künne, T.; Swarts, D.C.; Brouns, S.J. Planting the seed: Target recognition of short guide RNAs. Trends Microbiol. 2014, 22, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Josephs, E.A.; Kocak, D.D.; Fitzgibbon, C.J.; McMenemy, J.; Gersbach, C.A.; Marszalek, P.E. Structure and specificity of the RNA-guided endonuclease Cas9 during DNA interrogation, target binding and cleavage. Nucleic Acids Res. 2015, 43, 8924–8941. [Google Scholar] [CrossRef] [PubMed]
- Sipa, K.; Sochacka, E.; Kazmierczak-Baranska, J.; Maszewska, M.; Janicka, M.; Nowak, G.; Nawrot, B. Effect of base modifications on structure, thermodynamic stability, and gene silencing activity of short interfering RNA. Rna 2007, 13, 1301–1316. [Google Scholar] [CrossRef]
- Nazipova, N.N.; Shabalina, S.A. Understanding off-target effects through hybridization kinetics and thermodynamics. Cell Biol. Toxicol. 2020, 36, 11–15. [Google Scholar] [CrossRef]
- Xie, N.; Zhou, Y.; Sun, Q.; Tang, B. Novel epigenetic techniques provided by the CRISPR/Cas9 system. Stem Cells Int. 2018, 2018, 7834175. [Google Scholar] [CrossRef]
- Lo, A.; Qi, L. Genetic and epigenetic control of gene expression by CRISPR–Cas systems. F1000Research 2017, 6, Rev-747. [Google Scholar] [CrossRef]
- Enríquez, P. Focus: Epigenetics: CRISPR-mediated epigenome editing. Yale J. Biol. Med. 2016, 89, 471. [Google Scholar]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Fu, Y.; Reyon, D.; Joung, J.K. Targeted genome editing in human cells using CRISPR/Cas nucleases and truncated guide RNAs. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 546, pp. 21–45. [Google Scholar]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Ahkmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, H.; Lei, Y.-S.; Gong, C.-Y.; Chen, Z.; Luan, Y.-Q.; Li, Q.; Jian, Y.-Z.; Wang, H.-Z.; Wu, F.-L. Development of a self-restricting CRISPR-Cas9 system to reduce off-target effects. Mol. Ther.-Methods Clin. Dev. 2020, 18, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Greene, E.C. DNA repair pathway choices in CRISPR-Cas9-mediated genome editing. Trends Genet. 2021, 37, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Ata, H.; Ekstrom, T.L.; Martínez-Gálvez, G.; Mann, C.M.; Dvornikov, A.V.; Schaefbauer, K.J.; Ma, A.C.; Dobbs, D.; Clark, K.J.; Ekker, S.C. Robust activation of microhomology-mediated end joining for precision gene editing applications. PLoS Genet. 2018, 14, e1007652. [Google Scholar] [CrossRef]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Li, V.R.; Zhang, Z.; Troyanskaya, O.G. CROTON: An automated and variant-aware deep learning framework for predicting CRISPR/Cas9 editing outcomes. Bioinformatics 2021, 37, i342–i348. [Google Scholar] [CrossRef]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Chen, W.; McKenna, A.; Schreiber, J.; Haeussler, M.; Yin, Y.; Agarwal, V.; Noble, W.S.; Shendure, J. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 2019, 47, 7989–8003. [Google Scholar] [CrossRef]
- Leenay, R.T.; Aghazadeh, A.; Hiatt, J.; Tse, D.; Hultquist, J.F.; Krogan, N.; Wu, Z.; Marson, A.; May, A.P.; Zou, J. Systematic characterization of genome editing in primary T cells reveals proximal genomic insertions and enables machine learning prediction of CRISPR-Cas9 DNA repair outcomes. bioRxiv 2018, 404947. [Google Scholar] [CrossRef]
- Kosicki, M.; Allen, F.; Bradley, A. Cas9-induced large deletions and small indels are controlled in a convergent fashion. bioRxiv 2020. [Google Scholar] [CrossRef]
- Brinkman, E.K.; van Steensel, B. Rapid quantitative evaluation of CRISPR genome editing by TIDE and TIDER. In CRISPR Gene Editing; Springer: Berlin/Heidelberg, Germany, 2019; pp. 29–44. [Google Scholar]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, H.; Burger, A.; Biyong, B.; Felker, A.; Hess, C.; Zaugg, J.; Chiavacci, E.; Anders, C.; Jinek, M.; Mosimann, C. CrispRVariants charts the mutation spectrum of genome engineering experiments. Nat. Biotechnol. 2016, 34, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chang, H.Y.; Cho, S.W.; Ji, H.P. CRISPRpic: Fast and precise analysis for CRISPR-induced mutations via p refixed i ndex c ounting. NAR Genom. Bioinform. 2020, 2, lqaa012. [Google Scholar] [CrossRef] [PubMed]
- Güell, M.; Yang, L.; Church, G.M. Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 2014, 30, 2968–2970. [Google Scholar] [CrossRef]
- Park, J.; Lim, K.; Kim, J.-S.; Bae, S. Cas-analyzer: An online tool for assessing genome editing results using NGS data. Bioinformatics 2017, 33, 286–288. [Google Scholar] [CrossRef]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K. Inference of CRISPR edits from Sanger trace data. CRISPR J. 2022, 5, 123–130. [Google Scholar] [CrossRef]
- Yang, B.; Yang, L.; Chen, J. Development and application of base editors. CRISPR J. 2019, 2, 91–104. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, J.; Wei, P.; Zhang, B.; Gou, F.; Feng, Z.; Mao, Y.; Yang, L.; Zhang, H.; Xu, N. The CRISPR/C as9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 2014, 12, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Horlbeck, M.A.; Witkowsky, L.B.; Guglielmi, B.; Replogle, J.M.; Gilbert, L.A.; Villalta, J.E.; Torigoe, S.E.; Tjian, R.; Weissman, J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. elife 2016, 5, e12677. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Fløe, L.; Petersen, T.S.; Huang, J.; Xu, F.; Bolund, L.; Luo, Y.; Lin, L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017, 591, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Mäkelä, M.I.; Barker, H.R.; Bäuerlein, C.A.; Häkkinen, T.; Nykter, M.; Rämet, M. Chromatin accessibility is associated with CRISPR-Cas9 efficiency in the zebrafish (Danio rerio). PLoS ONE 2018, 13, e0196238. [Google Scholar] [CrossRef]
- Shin, J.; Jiang, F.; Liu, J.-J.; Bray, N.L.; Rauch, B.J.; Baik, S.H.; Nogales, E.; Bondy-Denomy, J.; Corn, J.E.; Doudna, J.A. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv. 2017, 3, e1701620. [Google Scholar] [CrossRef]
- Canver, M.C.; Joung, J.K.; Pinello, L. Impact of genetic variation on CRISPR-Cas targeting. CRISPR J. 2018, 1, 159–170. [Google Scholar] [CrossRef]
- Yan, J.; Chuai, G.; Zhou, C.; Zhu, C.; Yang, J.; Zhang, C.; Gu, F.; Xu, H.; Wei, J.; Liu, Q. Benchmarking CRISPR on-target sgRNA design. Brief. Bioinform. 2018, 19, 721–724. [Google Scholar] [CrossRef]


| Tool | Organism | Language | Cas Nucleases | Description | Database/ Web Server | Web Site (Accessed on 10 November 2022) | Ref. |
|---|---|---|---|---|---|---|---|
| CHOP CHOP | More than 100 | Python | Cas9, Cas12, Cas13 | Early web-based tool created by Harvard University to design gRNA based on matches and mismatches | Web server | http://crispor.tefor.net/ | [36] |
| CRISPR RGNE tools | More than 100 | Python | More than 20 Cas nucleases | Predicts multiple off-target and on-target effects based on the Cas-OFFinder model | Web server | http://www.rgenome.net/cas-designer/ | |
| CRISTA | More than 100 | Pearl and Python | Only Cas9 | Machine learning (ML)-based tools to predict off-target and on-target effects simultaneously | Web server | [35] | |
| GuideScan | Mouse and human | Python | Cpf1 and Cas9 | Predicts off-target effects based on sequence and structural features | Web server | https://guidescan.com/ | [37] |
| CRISPRDo | Human, mouse, zebrafish, and some worm species | Python | Cas9 and Cpf1 | Predict off-target and on-target effects simultaneously | Database | [38] | |
| sgRNACas9 | Mouse | Pearl | spCas9 | A web-based tool to predict off-target effects | Dataset | [39,40] | |
| EupaDGT | Eukaryotic pathogen | Python | More than 10 Cas nucleases | Machine-learning-based tool to predict on- and off-target effects simultaneously | Web-based | http://grna.ctegd.uga.edu/ | [41] |
| WU-CRISPR | Human and mouse | Pearl | SpCas9 | Machine-learning-algorithm-based tool that can predict off-target effects by providing sequences between 20 and 30,000 bp | Web-based | http://crispr.wustl.edu/ | [42] |
| CRISPR-P | 49 plant species | Python | More than 14 Cas nucleases orthologs | Web-based off-target and on-target prediction tools for a wide range of plant species | Web-based | http://crispr.hzau.edu.cn/CRISPR2 | [43] |
| CRISPRz | Zebrafish, human, and mouse | Python | spCas9 | Trained on large datasets from zebrafish, humans, and mice to generate a gRNA dataset | Web-based | https://research.nhgri.nih.gov/CRISPRz/ | [44] |
| PhytoCRISPR x | Wide range of plant species and especially phytoplankton | Pearl/bash | SpCas9. Cas12 | Web-based tool to predict off-target effects | Dataset | [46] | |
| CRISPRPOR | More than 100 species | Python | More than 30 Cas orthologs | Design gRNA dataset based on match/match in seed regions | Web tool | http://crispor.tefor.net/ | [45] |
| Png Designer | 6 | Python | Cas9 | A newly designed tool for generating guide RNA for base editing and prime editing | Web tool | https://www.crisprindelphi.design/ | [47] |
| Model | Repair Prediction | Cell Lines | Remarks | Web Site (Accessed on 10 November 2022) | Ref. |
|---|---|---|---|---|---|
| FORECast | Can predict deletions as well as insertions, with 420 and 20 classes, respectively | iPSC, CHOHAP1, mESCs, K562, and RPE1 | Created through multi-class logistic regression | https://partslab.sanger.ac.uk/FORECasT | [66] |
| CROTON | K562 | Can predict the in-frameshift frequency with 1/2 bp | Created through CNN+ NAS | https://github.com/vli31/CROTON | [67] |
| InDelphi | HEK293, K562, HCT116, mESCs, and U20S | Can predict microhomology deletions (90 classes), non-microhomology deletions (59 classes), and 4 classes of 1 bp insertions | Generated through deep neural network and K-Nearest Neighbor | https://indelphi.giffordlab.mit.edu/about | [68] |
| Lindel | HEK293T | Can predict deletions (536 classes) and insertions (21 classes) | Generated through logistic regression | https://lindel.gs.washington.edu/Lindel | [69] |
| SPROUT | T cells | Can predict repairs such as INDELS | Gradient Boosting Decision Tree | https://zou-group.github.io/SPROUT | [70] |
| Apindel | K562 | Can predict insertions (536 classes) and deletions (21 classes) | Glove + Positional Encoding | [71] |
| Tools | Analysis Basis | Input | Output | Supported Experiment | Supported Cas9 Nucleases | Ref. |
|---|---|---|---|---|---|---|
| CRISPResso2 | NGS | FASTQ | Sequence alignment, NHEJ/HDR events | For CRISPR/Cas9, base editors | Cas9, Cpf1 | [78] |
| CasAnalyzer | NGS | FASTQ | Sequence alignment, HDR/NHEJ events | CRISPR/Cas9 | spCas9, StCas9, HFCas9, SaCas9, Cpf1, CjCas9 | [77] |
| CRISPR-GA | NGS | FASTQ | INDEL frequency, recombination due to HRD events | Only Cas9 | Cas9 | [76] |
| TIDE/TIDER | Sanger | ABI | INDELS/HDR frequency | Only CRISPR | spCas9, SaCas9, FnCas9, AsCpf1, stCas9 | [72] |
| ICE | Sanger | ABI | INDELS/HDR frequency | Only CRISPR/Cas9 | Cas9, SaCas9 | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naeem, M.; Alkhnbashi, O.S. Current Bioinformatics Tools to Optimize CRISPR/Cas9 Experiments to Reduce Off-Target Effects. Int. J. Mol. Sci. 2023, 24, 6261. https://doi.org/10.3390/ijms24076261
Naeem M, Alkhnbashi OS. Current Bioinformatics Tools to Optimize CRISPR/Cas9 Experiments to Reduce Off-Target Effects. International Journal of Molecular Sciences. 2023; 24(7):6261. https://doi.org/10.3390/ijms24076261
Chicago/Turabian StyleNaeem, Muhammad, and Omer S. Alkhnbashi. 2023. "Current Bioinformatics Tools to Optimize CRISPR/Cas9 Experiments to Reduce Off-Target Effects" International Journal of Molecular Sciences 24, no. 7: 6261. https://doi.org/10.3390/ijms24076261