
Changes in Memory, Sedation, and Receptor Kinetics Imparted by the β2-N265M and β3-N265M GABAA Receptor Point Mutations

Abstract
:1. Introduction
2. Results
2.1. Behavioral Characteristics of β2-N265M and β3-N265M Mice
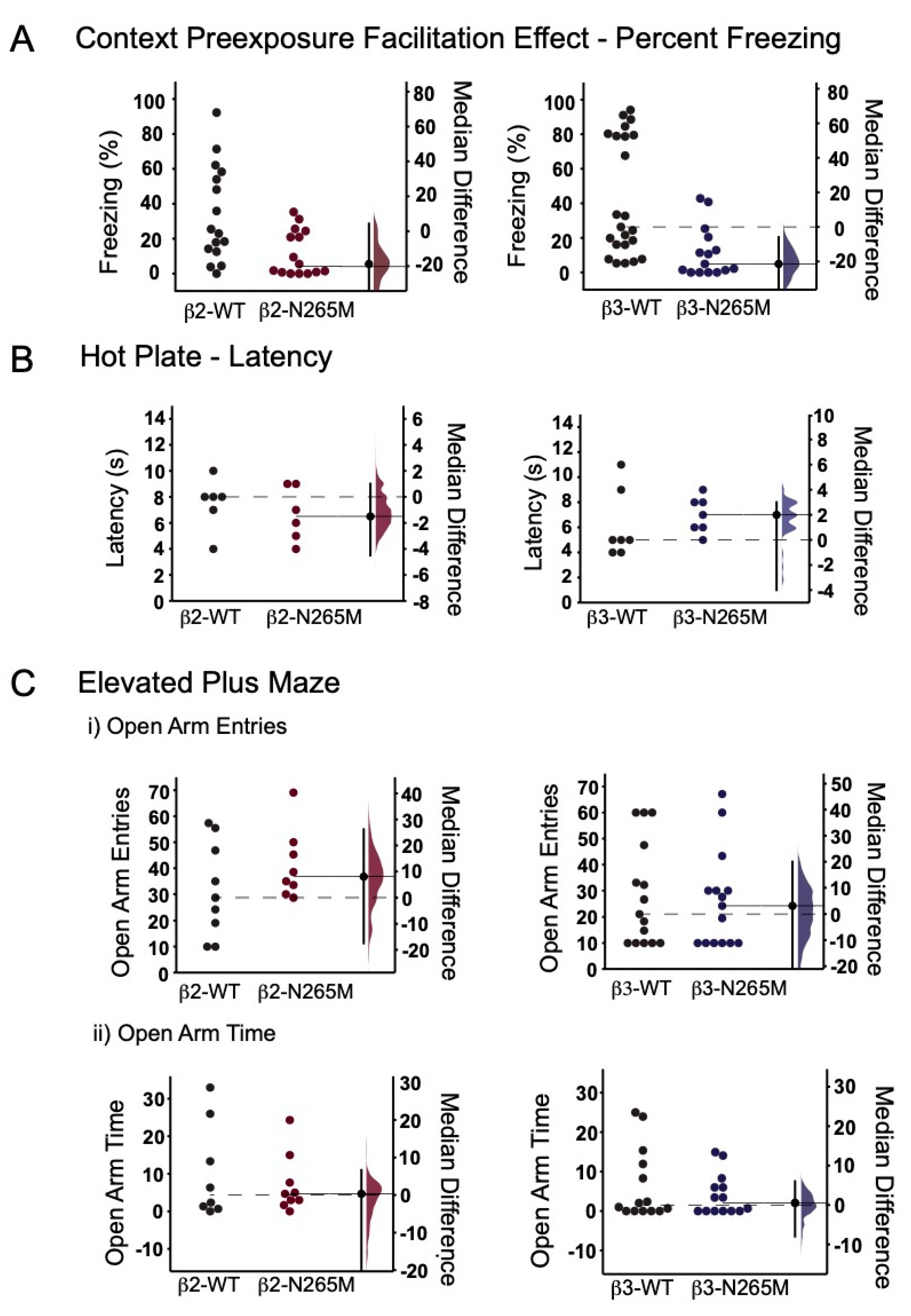
2.1.1. CPFE Learning Test
2.1.2. Hotplate Test
2.1.3. Elevated Plus Maze Test
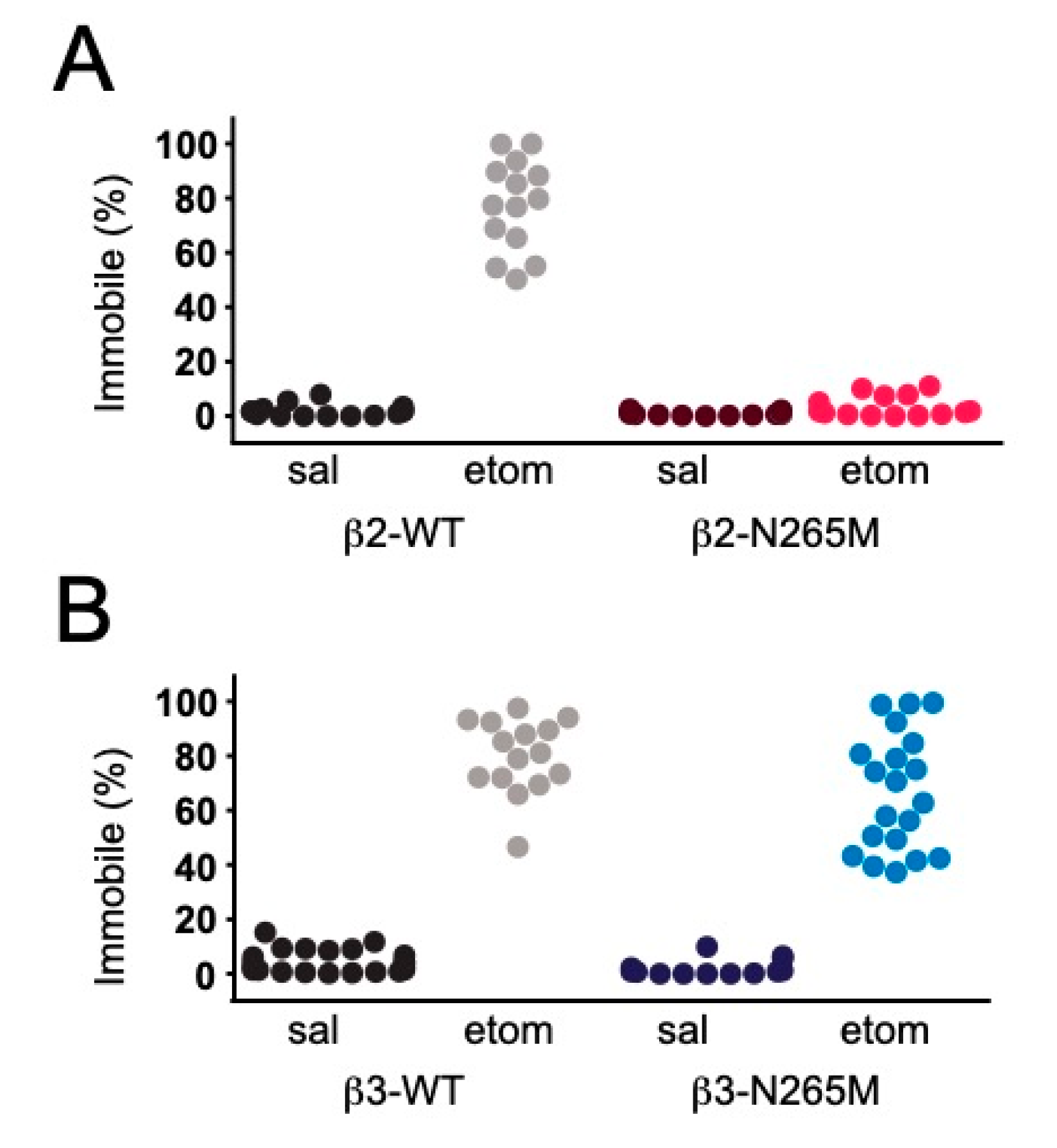
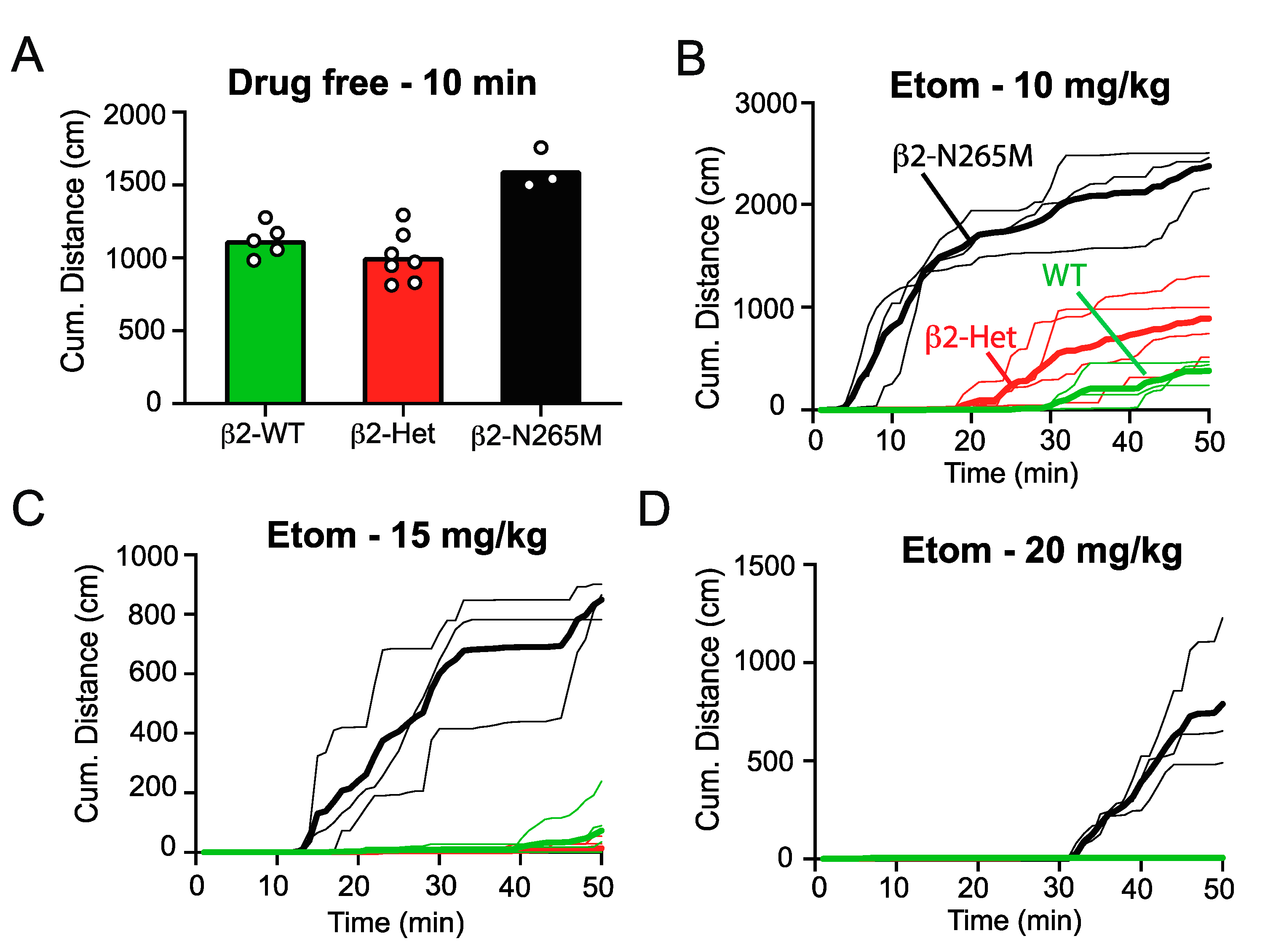
2.1.4. Sedation
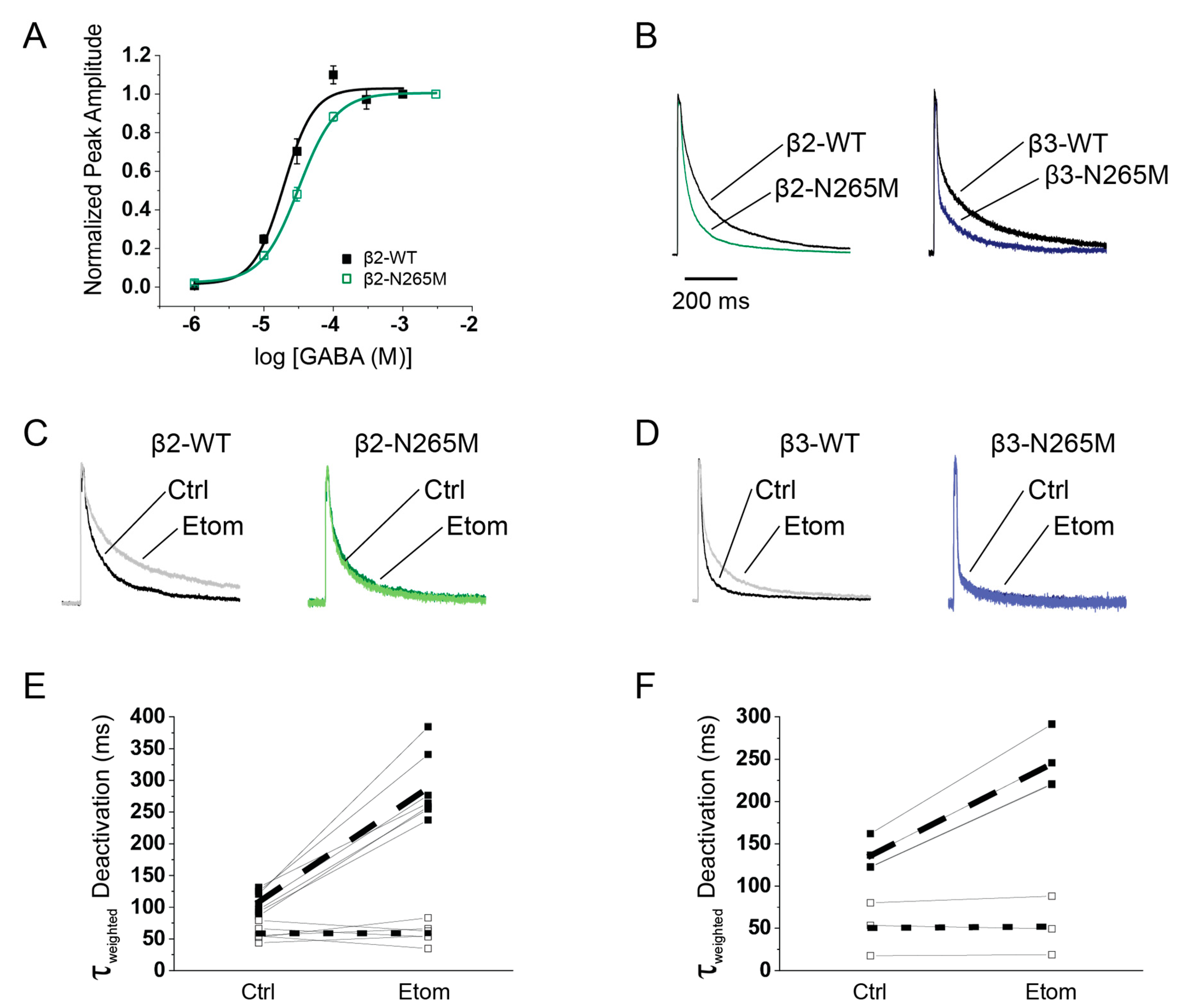
2.2. Expressed Receptor Characteristics
3. Discussion
4. Materials and Methods
4.1. Experimental Mice
4.2. Genotyping
4.3. Behavioral Studies
4.3.1. Context Preexposure Facilitation Effect (CPFE)
4.3.2. Elevated Plus Maze (EPM) Test
4.3.3. Hotplate Test
4.3.4. Open Field Test (OFT)
4.4. Recombinant Receptor Recordings
4.5. Data Presentation and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hemmings, H.C.; Riegelhaupt, P.M.; Kelz, M.B.; Solt, K.; Eckenhoff, R.G.; Orser, B.A.; Goldstein, P.A. Towards a Comprehensive Understanding of Anesthetic Mechanisms of Action: A Decade of Discovery. Trends Pharmacol. Sci. 2019, 40, 464–481. [Google Scholar] [CrossRef]
- Garcia, P.S.; Kolesky, S.E.; Jenkins, A. General anesthetic actions on GABA(A) receptors. Curr. Neuropharmacol. 2010, 8, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Franks, N.P. General anaesthesia: From molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 2008, 9, 370–386. [Google Scholar] [CrossRef] [PubMed]
- Antkowiak, B.; Rammes, G. GABA(A) receptor-targeted drug development -New perspectives in perioperative anesthesia. Expert Opin. Drug Discov. 2019, 14, 683–699. [Google Scholar] [CrossRef]
- Weir, C.J.; Mitchell, S.J.; Lambert, J.J. Role of GABA(A) receptor subtypes in the behavioural effects of intravenous general anaesthetics. Br. J. Anaesth. 2017, 119, I167–I175. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Olsen, R.W. GABA A receptor channels. Annu. Rev. Neurosci. 1994, 17, 569–602. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W.; Sieghart, W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: Classification on the basis of subunit composition, pharmacology, and function. Update. Pharm. Rev. 2008, 60, 243–260. [Google Scholar] [CrossRef]
- Olsen, R.W. GABA(A) receptor: Positive and negative allosteric modulators. Neuropharmacology 2018, 136, 10–22. [Google Scholar] [CrossRef]
- Mody, I.; Pearce, R.A. Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 2004, 27, 569–575. [Google Scholar] [CrossRef]
- Rudolph, U.; Crestani, F.; Benke, D.; Brunig, I.; Benson, J.A.; Fritschy, J.M.; Martin, J.R.; Bluethmann, H.; Mohler, H. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature 1999, 401, 796–800. [Google Scholar] [CrossRef]
- Low, K.; Crestani, F.; Keist, R.; Benke, D.; Brunig, I.; Benson, J.A.; Fritschy, J.M.; Rulicke, T.; Bluethmann, H.; Mohler, H.; et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science 2000, 290, 131–134. [Google Scholar] [CrossRef]
- Smith, K.S.; Engin, E.; Meloni, E.G.; Rudolph, U. Benzodiazepine-induced anxiolysis and reduction of conditioned fear are mediated by distinct GABAA receptor subtypes in mice. Neuropharmacology 2012, 63, 250–258. [Google Scholar] [CrossRef]
- Morris, H.V.; Dawson, G.R.; Reynolds, D.S.; Atack, J.R.; Stephens, D.N. Both alpha2 and alpha3 GABAA receptor subtypes mediate the anxiolytic properties of benzodiazepine site ligands in the conditioned emotional response paradigm. Eur. J. Neurosci. 2006, 23, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.Y.; Martin, L.J.; Elliott, E.M.; Kim, J.H.; Mount, H.T.; Taverna, F.A.; Roder, J.C.; MacDonald, J.F.; Bhambri, A.; Collinson, N.; et al. Alpha5GABAA receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate. J. Neurosci. 2006, 26, 3713–3720. [Google Scholar] [CrossRef]
- Rudolph, U.; Mohler, H. GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 483–507. [Google Scholar] [CrossRef]
- Belelli, D.; Lambert, J.J.; Peters, J.A.; Wafford, K.; Whiting, P.J. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. USA 1997, 94, 11031–11036. [Google Scholar] [CrossRef]
- Krasowski, M.D.; Koltchine, V.V.; Rick, C.E.; Ye, Q.; Finn, S.E.; Harrison, N.L. Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyric acid type A receptor distinct from that for isoflurane. Mol. Pharmacol. 1998, 53, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.S.; Rosahl, T.W.; Cirone, J.; O’Meara, G.F.; Haythornthwaite, A.; Newman, R.J.; Myers, J.; Sur, C.; Howell, O.; Rutter, A.R.; et al. Sedation and anesthesia mediated by distinct GABA(A) receptor isoforms. J. Neurosci. 2003, 23, 8608–8617. [Google Scholar] [CrossRef] [PubMed]
- Zeller, A.; Arras, M.; Lazaris, A.; Jurd, R.; Rudolph, U. Distinct molecular targets for the central respiratory and cardiac actions of the general anesthetics etomidate and propofol. FASEB J. 2005, 19, 1677–1679. [Google Scholar] [CrossRef]
- Jurd, R.; Arras, M.; Lambert, S.; Drexler, B.; Siegwart, R.; Crestani, F.; Zaugg, M.; Vogt, K.E.; Ledermann, B.; Antkowiak, B.; et al. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003, 17, 250–252. [Google Scholar] [CrossRef]
- Zarnowska, E.D.; Rodgers, F.C.; Oh, I.; Rau, V.; Lor, C.; Laha, K.T.; Jurd, R.; Rudolph, U.; Eger, E.I.N.; Pearce, R.A. Etomidate blocks LTP and impairs learning but does not enhance tonic inhibition in mice carrying the N265M point mutation in the beta3 subunit of the GABA(A) receptor. Neuropharmacology 2015, 93, 171–178. [Google Scholar] [CrossRef]
- Figueroa, A.G.; Benkwitz, C.; Surges, G.; Kunz, N.; Homanics, G.E.; Pearce, R.A. Hippocampal beta2-GABAA receptors mediate LTP suppression by etomidate and contribute to long-lasting feedback but not feedforward inhibition of pyramidal neurons. J. Neurophysiol. 2021, 126, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Sonner, J.M.; Jurd, R.; Rudolph, U.; Borghese, C.M.; Harris, R.A.; Laster, M.J.; Eger, E.I. Beta3-containing gamma-aminobutyric acidA receptors are not major targets for the amnesic and immobilizing actions of isoflurane. Anesth. Analg. 2005, 101, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Zeller, A.; Arras, M.; Jurd, R.; Rudolph, U. Mapping the contribution of beta3-containing GABAA receptors to volatile and intravenous general anesthetic actions. BMC. Pharmacol. 2007, 7, 2. [Google Scholar] [CrossRef]
- Drexler, B.; Jurd, R.; Rudolph, U.; Antkowiak, B. Distinct actions of etomidate and propofol at beta3-containing gamma-aminobutyric acid type A receptors. Neuropharmacology 2009, 57, 446–455. [Google Scholar] [CrossRef]
- Siegwart, R.; Jurd, R.; Rudolph, U. Molecular determinants for the action of general anesthetics at recombinant alpha(2)beta(3)gamma(2) gamma-aminobutyric acid(A) receptors. J. Neurochem. 2002, 80, 140–148. [Google Scholar] [CrossRef]
- Burkat, P.M.; Lor, C.; Perouansky, M.; Pearce, R.A. Enhancement of alpha5-containing gamma-aminobutyric acid type A receptors by the nonimmobilizer 1,2-dichlorohexafluorocyclobutane (F6) is abolished by the beta3(N265M) mutation. Anesth. Analg. 2014, 119, 1277–1284. [Google Scholar] [CrossRef]
- Siegwart, R.; Krahenbuhl, K.; Lambert, S.; Rudolph, U. Mutational analysis of molecular requirements for the actions of general anaesthetics at the gamma-aminobutyric acidA receptor subtype, alpha1beta2gamma2. BMC. Pharmacol. 2003, 3, 13. [Google Scholar] [CrossRef]
- Desai, R.; Ruesch, D.; Forman, S.A. Gamma-amino butyric acid type A receptor mutations at beta2N265 alter etomidate efficacy while preserving basal and agonist-dependent activity. Anesthesiology 2009, 111, 774–784. [Google Scholar] [CrossRef]
- Jacob, T.C. Neurobiology and Therapeutic Potential of alpha5-GABA Type A Receptors. Front. Mol. Neurosci. 2019, 12, 179. [Google Scholar] [CrossRef]
- Zecharia, A.Y.; Nelson, L.E.; Gent, T.C.; Schumacher, M.; Jurd, R.; Rudolph, U.; Brickley, S.G.; Maze, M.; Franks, N.P. The involvement of hypothalamic sleep pathways in general anesthesia: Testing the hypothesis using the GABAA receptor beta3N265M knock-in mouse. J. Neurosci. 2009, 29, 2177–2187. [Google Scholar] [CrossRef]
- Drexler, B.; Jurd, R.; Rudolph, U.; Antkowiak, B. Dual actions of enflurane on postsynaptic currents abolished by the gamma-aminobutyric acid type A receptor beta3(N265M) point mutation. Anesthesiology 2006, 105, 297–304. [Google Scholar] [CrossRef]
- Perouansky, M. Coagulation, flocculation, and denaturation: A century of research into protoplasmic theories of anesthesia. Anesth. Analg. 2014, 119, 311–320. [Google Scholar] [CrossRef]
- Franks, N.P.; Lieb, W.R. Molecular and cellular mechanisms of general anaesthesia. Nature 1994, 367, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Pittson, S.; Himmel, A.M.; MacIver, M.B. Multiple synaptic and membrane sites of anesthetic action in the CA1 region of rat hippocampal slices. BMC Neurosci. 2004, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Sonner, J.M.; Husain, S.S.; Miller, K.W.; Jurd, R.; Rudolph, U.; Eger, E.I. R (+) etomidate and the photoactivable R (+) azietomidate have comparable anesthetic activity in wild-type mice and comparably decreased activity in mice with a N265M point mutation in the gamma-aminobutyric acid receptor beta3 subunit. Anesth. Analg. 2005, 101, 131–135. [Google Scholar] [CrossRef]
- Pistis, M.; Belelli, D.; McGurk, K.; Peters, J.A.; Lambert, J.J. Complementary regulation of anaesthetic activation of human (alpha6beta3gamma2L) and Drosophila (RDL) GABA receptors by a single amino acid residue. J. Physiol. 1999, 515, 3–18. [Google Scholar] [CrossRef]
- Jones, M.V.; Sahara, Y.; Dzubay, J.A.; Westbrook, G.L. Defining affinity with the GABAA receptor. J. Neurosci. 1998, 18, 8590–8604. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.T.; Macdonald, R.L. Agonist Trapping by GABAA Receptor Channels. J. Neurosci. 2001, 21, 9083–9091. [Google Scholar] [CrossRef]
- McKinstry-Wu, A.R.; Wasilczuk, A.Z.; Dailey, W.P.; Eckenhoff, R.G.; Kelz, M.B. In Vivo Photoadduction of Anesthetic Ligands in Mouse Brain Markedly Extends Sedation and Hypnosis. J. Neurosci. 2023. [Google Scholar] [CrossRef]
- Reitz, S.L.; Kelz, M.B. Preoptic Area Modulation of Arousal in Natural and Drug Induced Unconscious States. Front. Neurosci. 2021, 15, 644330. [Google Scholar] [CrossRef]
- Prevot, T.; Sibille, E. Altered GABA-mediated information processing and cognitive dysfunctions in depression and other brain disorders. Mol. Psychiatry 2020, 26, 151–167. [Google Scholar] [CrossRef]
- Elgersma, Y.; Sweatt, J.D.; Giese, K.P. Mouse genetic approaches to investigating calcium/calmodulin-dependent protein kinase II function in plasticity and cognition. J. Neurosci. 2004, 24, 8410–8415. [Google Scholar] [CrossRef] [PubMed]
- Zarnowska, E.D.; Rudolph, U.; Pearce, R.A. GABAa,slow IPSCs result from neurotransmitter spillover onto extrasynaptic receptors containing a5 subunits. Soc. Neurosci. Abstr. 2006. [Google Scholar]
- Sperk, G.; Schwarzer, C.; Tsunashima, K.; Fuchs, K.; Sieghart, W. GABA(A) receptor subunits in the rat hippocampus I: Immunocytochemical distribution of 13 subunits. Neuroscience 1997, 80, 987–1000. [Google Scholar] [CrossRef]
- Olsen, R.W.; Sieghart, W. GABA(A) receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar] [CrossRef]
- Zhu, M.; Abdulzahir, A.; Perkins, M.G.; Krause, B.M.; Lennertz, R.; Ruhl, D.; Hentschke, H.; Nagarajan, R.; Chapman, E.R.; Rudolph, U.; et al. Control of contextual memory through interneuronal alpha5-GABAA receptors. BioRxiv 2022. [Google Scholar] [CrossRef]
- Deng, Y.; Bi, M.; Delerue, F.; Forrest, S.L.; Chan, G.; van der Hoven, J.; van Hummel, A.; Feiten, A.F.; Lee, S.; Martinez-Valbuena, I.; et al. Loss of LAMP5 interneurons drives neuronal network dysfunction in Alzheimer’s disease. Acta Neuropathol. 2022, 144, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, M.; Han, Y.; Zhang, H. GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment. Front. Neurosci. 2020, 14, 660. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Borghese, C.M.; Ruiz, C.I.; Cullins, M.A.; Da Costa, A.; Osterndorff-Kahanek, E.A.; Homanics, G.E.; Harris, R.A. Mutation of the inhibitory ethanol site in GABAA rho1 receptors promotes tolerance to ethanol-induced motor incoordination. Neuropharmacology 2017, 123, 201–209. [Google Scholar] [CrossRef]
- Everington, E.A.; Gibbard, A.G.; Swinny, J.D.; Seifi, M. Molecular Characterization of GABA-A Receptor Subunit Diversity within Major Peripheral Organs and Their Plasticity in Response to Early Life Psychosocial Stress. Front. Mol. Neurosci. 2018, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Homanics, G.E.; Delorey, T.M.; Firestone, L.L.; Quinlan, J.J.; Handforth, A.; Harrison, N.L.; Krasowski, M.D.; Rick, C.E.; Korpi, E.R.; Makela, R.; et al. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc. Natl. Acad. Sci. USA 1997, 94, 4143–4148. [Google Scholar] [CrossRef]
- Fanselow, M.S. Factors governing one-trial contextual conditioning. Anim. Learn. Behav. 1990, 18, 264–270. [Google Scholar] [CrossRef]
- Rudy, J.W. Context representations, context functions, and the parahippocampal-hippocampal system. Learn. Mem. 2009, 16, 573–585. [Google Scholar] [CrossRef]
- Pinizzotto, C.C.; Heroux, N.A.; Horgan, C.J.; Stanton, M.E. Role of dorsal and ventral hippocampal muscarinic receptor activity in acquisition and retention of contextual fear conditioning. Behav. Neurosci. 2020, 134, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.A.; Heroux, N.A.; Stanton, M.E. NMDA receptors and the ontogeny of post-shock and retention freezing during contextual fear conditioning. Dev. Psychobiol. 2020, 62, 380–385. [Google Scholar] [CrossRef]
- Krasne, F.B.; Cushman, J.D.; Fanselow, M.S. A Bayesian context fear learning algorithm/automaton. Front. Behav. Neurosci. 2015, 9, 112. [Google Scholar] [CrossRef]
- Fanselow, M.S. Contextual fear, gestalt memories, and the hippocampus. Behav. Brain Res. 2000, 110, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Tumkaya, T.; Aryal, S.; Choi, H.; Claridge-Chang, A. Moving beyond P values: Data analysis with estimation graphics. Nat. Methods 2019, 16, 565–566. [Google Scholar] [CrossRef]
- Calin-Jageman, R.J.; Cumming, G. Estimation for Better Inference in Neuroscience. Eneuro 2019, 6. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | ||||
|---|---|---|---|---|
| Current Phase | α5β2γ2L | α5β2(N265M)γ2L | α5β3γ2L | α5β3(N265M)γ2L |
| 10–90% Activation (ms) | 1.20 ± 0.13 | 1.6 ± 0.7 * | 1.66 ± 0.17 | 2.08 ± 0.28 |
| τfast Deactivation (ms) | 41 ± 3.4 | 22 ± 2.1 *** | 21.0 ± 6.4 | 3.73 ± 0.42 *** |
| Fraction Fast | 0.60 ± 0.02 | 0.68 ± 0.02 * | 0.60 ± 0.08 | 0.67 ± 0.07 |
| τslow Deactivation (ms) | 211 ± 22 | 137 ± 14 * | 225 ± 33 | 91.4 ± 12.8 ** |
| Fraction Slow | 0.4 ± 0.02 | 0.32 ± 0.02 * | 0.4 ± 0.08 | 0.33 ± 0.07 |
| τweighted Deactivation (ms) | 107 ± 9.6 | 60 ± 7.0 ** | 114 ± 27 | 36.7 ± 10.0 * |
| τweighted with 1 μM etomidate | 285 ± 42 | 71.4 ± 9.6 *** | 214 ± 17 | 37.7 ± 1.9 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdulzahir, A.; Klein, S.; Lor, C.; Perkins, M.G.; Frelka, A.; Pearce, R.A. Changes in Memory, Sedation, and Receptor Kinetics Imparted by the β2-N265M and β3-N265M GABAA Receptor Point Mutations. Int. J. Mol. Sci. 2023, 24, 5637. https://doi.org/10.3390/ijms24065637
Abdulzahir A, Klein S, Lor C, Perkins MG, Frelka A, Pearce RA. Changes in Memory, Sedation, and Receptor Kinetics Imparted by the β2-N265M and β3-N265M GABAA Receptor Point Mutations. International Journal of Molecular Sciences. 2023; 24(6):5637. https://doi.org/10.3390/ijms24065637
Chicago/Turabian StyleAbdulzahir, Alifayaz, Steven Klein, Chong Lor, Mark G. Perkins, Alyssa Frelka, and Robert A. Pearce. 2023. "Changes in Memory, Sedation, and Receptor Kinetics Imparted by the β2-N265M and β3-N265M GABAA Receptor Point Mutations" International Journal of Molecular Sciences 24, no. 6: 5637. https://doi.org/10.3390/ijms24065637