
RPflex: A Coarse-Grained Network Model for RNA Pocket Flexibility Study

Abstract
:1. Introduction
2. Results
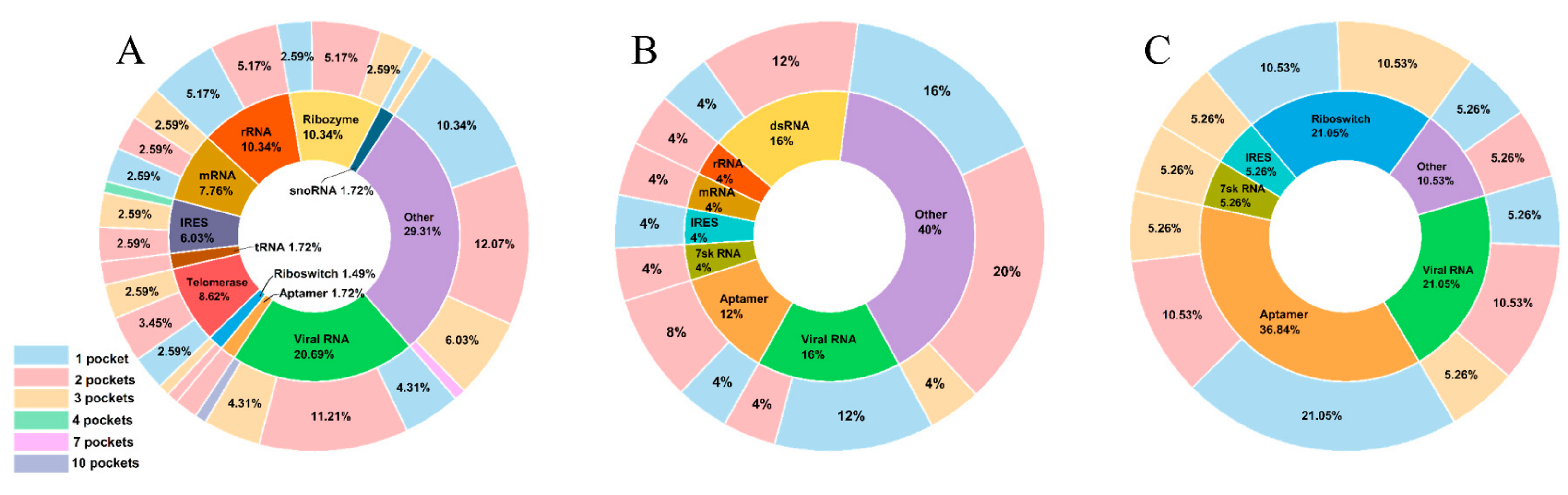
2.1. Overview of the RNA Pocket Dataset
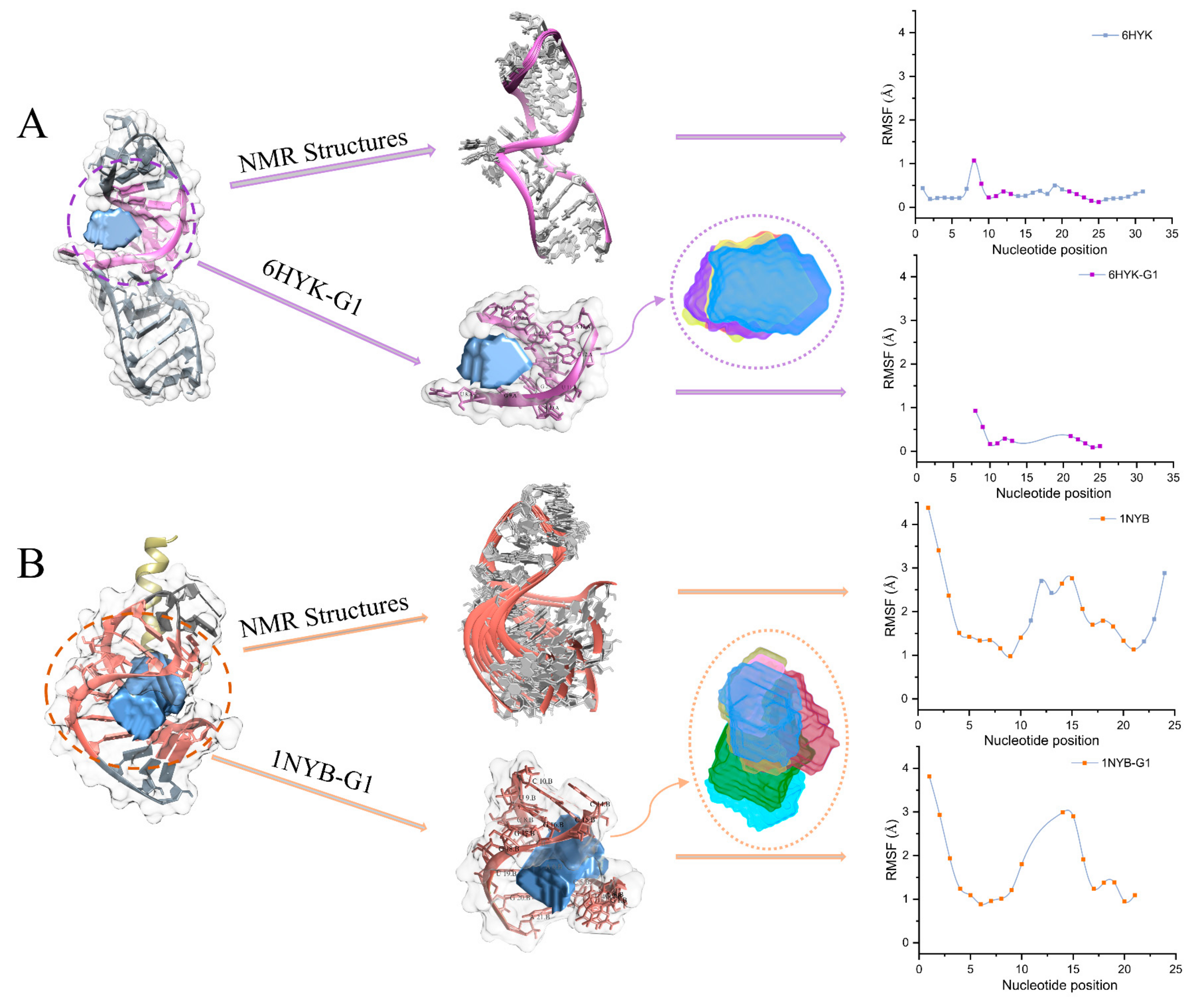
2.2. RMSF Analysis of Pockets
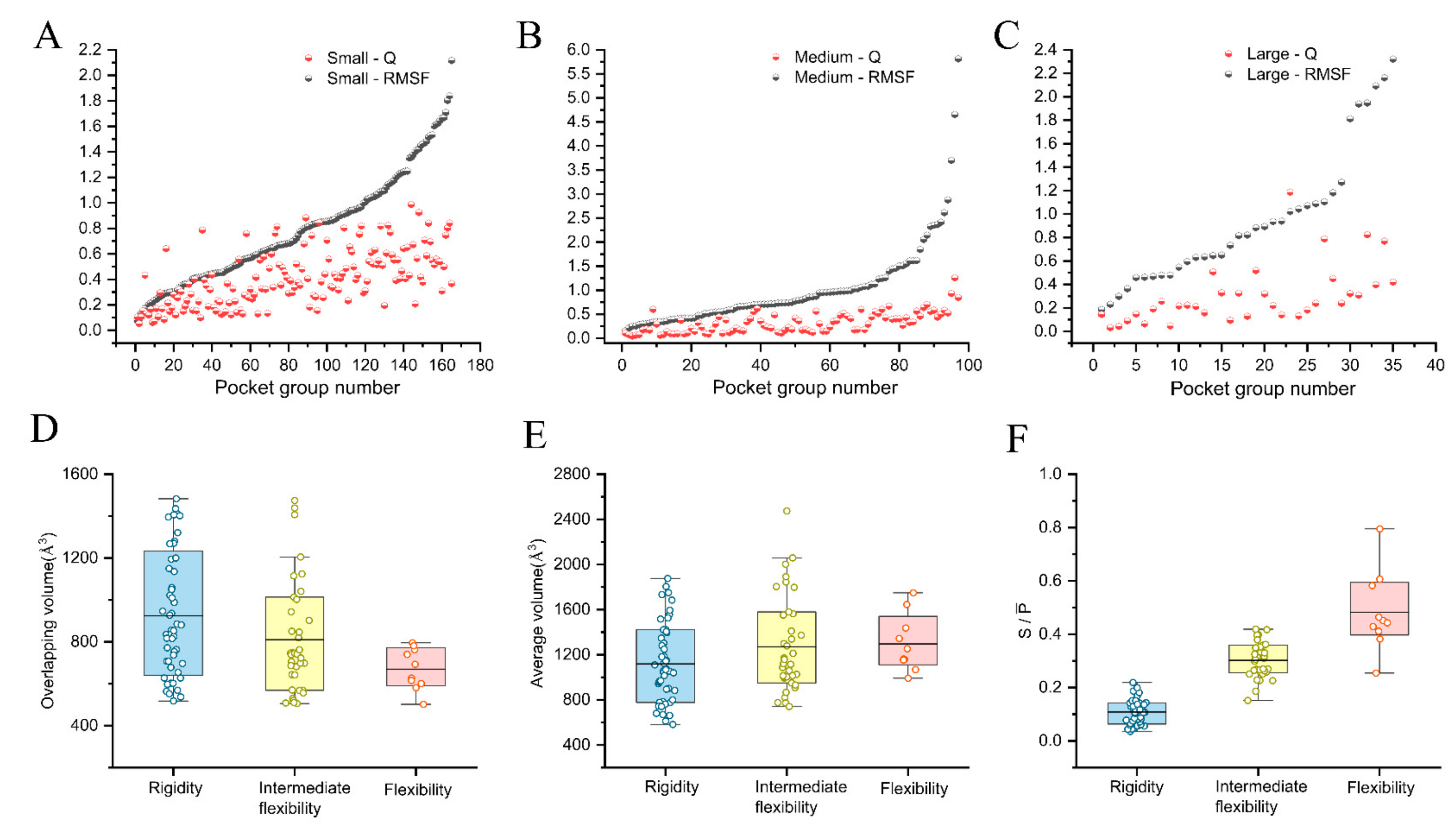
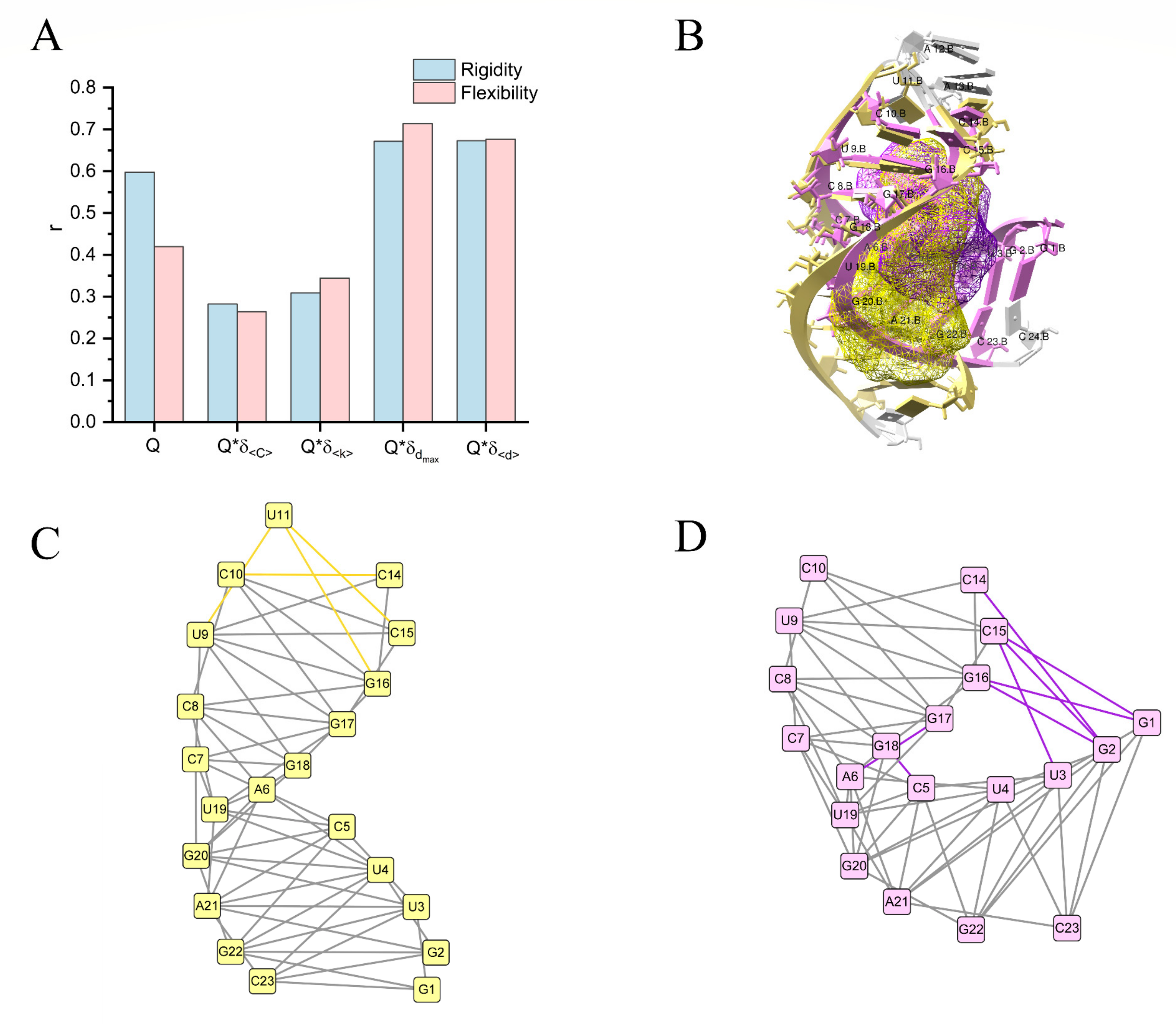
2.3. Quantitative Analysis of the Pocket Flexibility
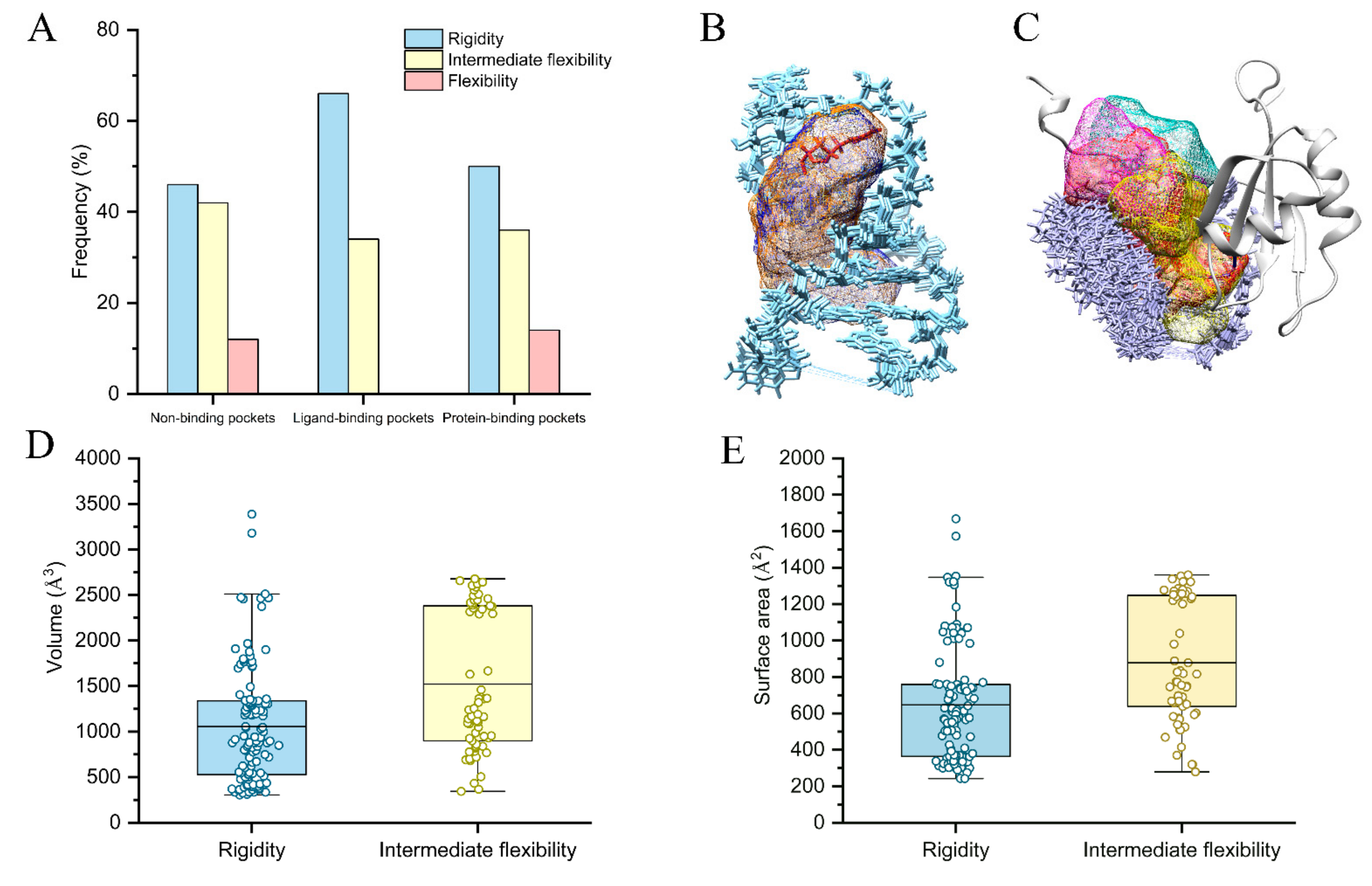
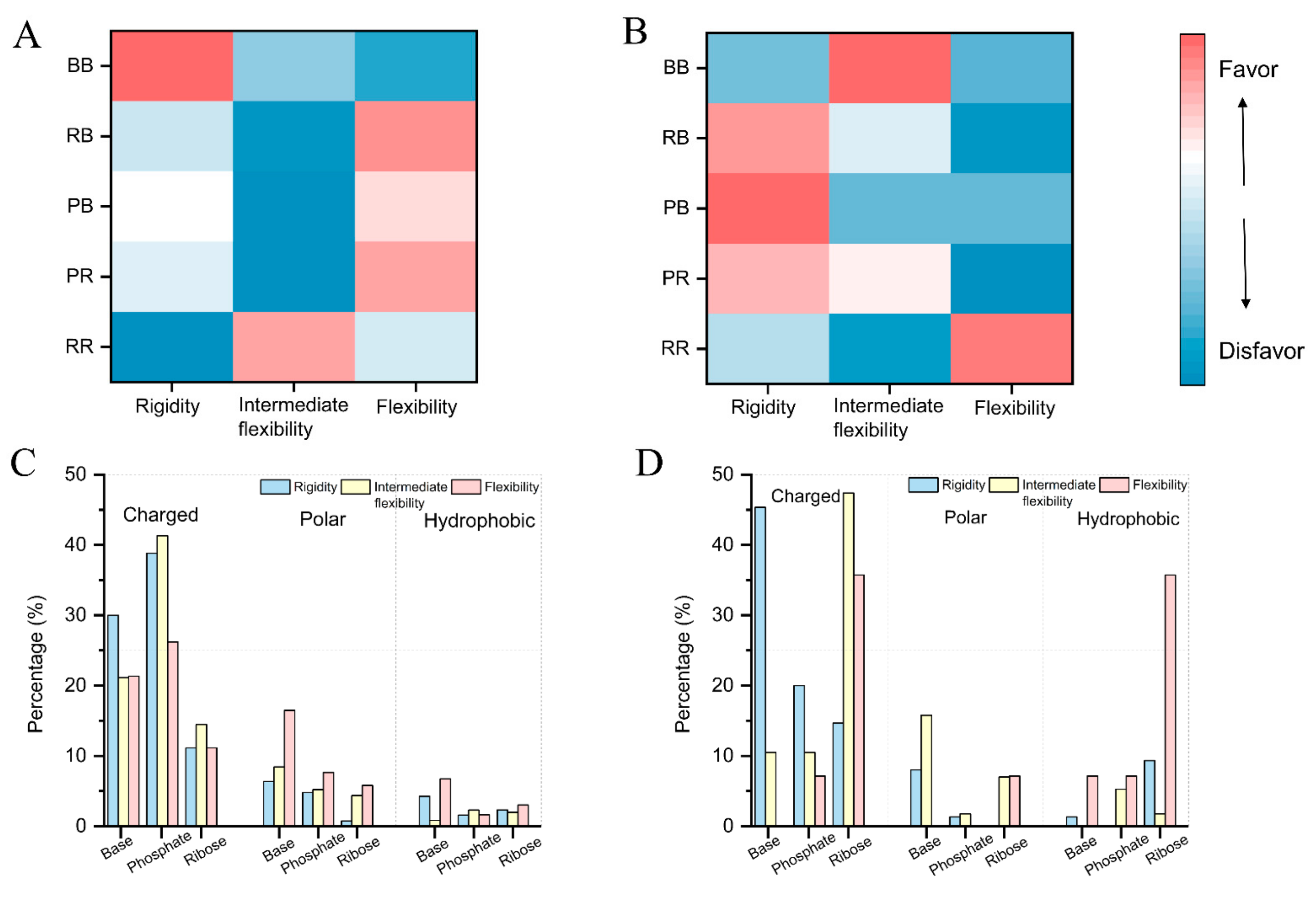
2.4. Flexibility on Binding and Unbinding Pockets
2.5. Physics-Based Interactions on Flexibility
3. Discussion
4. Materials and Methods
4.1. Structure Dataset Collection
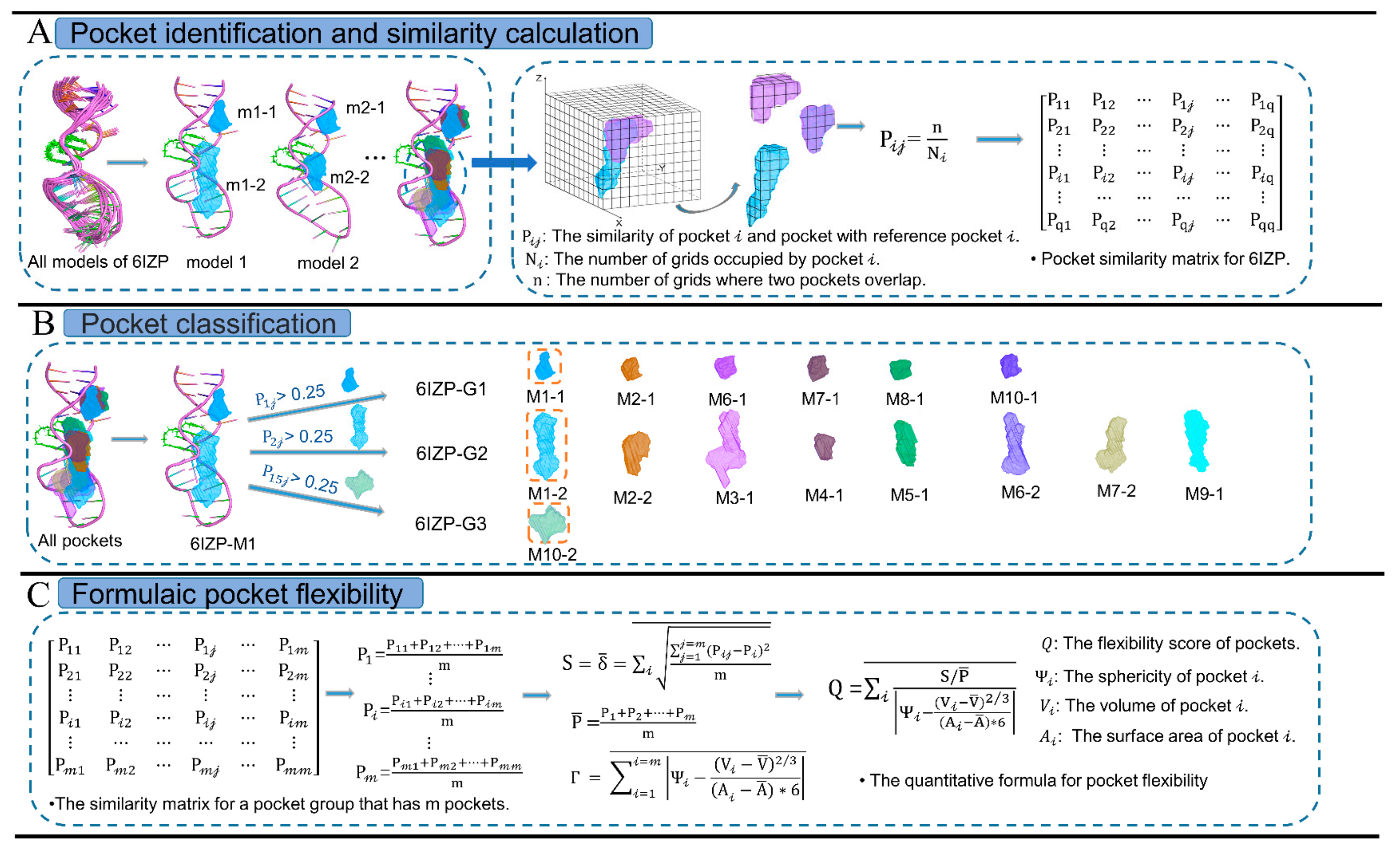
4.2. Criteria for Pocket Conformational Flexibility
4.3. Network Construction
4.4. RMSF Calculation
4.5. Chemical Group and Interaction Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Huang, L.; Patel, D.J. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature 2009, 458, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef]
- Howe, J.A.; Wang, H.; Fischmann, T.O.; Balibar, C.J.; Xiao, L.; Galgoci, A.M.; Malinverni, J.C.; Mayhood, T.; Villafania, A.; Nahvi, A.; et al. Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526, 672–677. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020, 182, 417–428.e413. [Google Scholar] [CrossRef]
- Haller, A.; Souliere, M.F.; Micura, R. The dynamic nature of RNA as key to understanding riboswitch mechanisms. Acc. Chem. Res. 2011, 44, 1339–1348. [Google Scholar] [CrossRef]
- Umuhire Juru, A.; Patwardhan, N.N.; Hargrove, A.E. Understanding the Contributions of Conformational Changes, Thermodynamics, and Kinetics of RNA-Small Molecule Interactions. ACS Chem. Biol. 2019, 14, 824–838. [Google Scholar] [CrossRef]
- Ganser, L.R.; Kelly, M.L.; Herschlag, D.; Al-Hashimi, H.M. The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol. 2019, 20, 474–489. [Google Scholar] [CrossRef]
- Haque, F.; Pi, F.; Zhao, Z.; Gu, S.; Hu, H.; Yu, H.; Guo, P. RNA versatility, flexibility, and thermostability for practice in RNA nanotechnology and biomedical applications. Wiley Interdiscip. Rev. RNA 2018, 9, e1452. [Google Scholar] [CrossRef]
- Nam, Y.; Chen, C.; Gregory, R.I.; Chou, J.J.; Sliz, P. Molecular basis for interaction of let-7 microRNAs with Lin28. Cell 2011, 147, 1080–1091. [Google Scholar] [CrossRef] [Green Version]
- Parsons, J.; Castaldi, M.P.; Dutta, S.; Dibrov, S.M.; Wyles, D.L.; Hermann, T. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat. Chem. Biol. 2009, 5, 823–825. [Google Scholar] [CrossRef]
- Turnbull, A.P.; Wu, X. Studying RNA-Protein Complexes Using X-Ray Crystallography. Methods Mol. Biol. 2021, 2263, 423–446. [Google Scholar] [CrossRef]
- Bothe, J.R.; Nikolova, E.N.; Eichhorn, C.D.; Chugh, J.; Hansen, A.L.; Al-Hashimi, H.M. Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy. Nat. Methods 2011, 8, 919–931. [Google Scholar] [CrossRef]
- Barnwal, R.P.; Yang, F.; Varani, G. Applications of NMR to structure determination of RNAs large and small. Arch. Biochem. Biophys. 2017, 628, 42–56. [Google Scholar] [CrossRef]
- Zeng, C.; Jian, Y.; Vosoughi, S.; Zeng, C.; Zhao, Y. Evaluating native-like structures of RNA-protein complexes through the deep learning method. Nat. Commun. 2023, 14, 1060. [Google Scholar] [CrossRef]
- Madan, B.; Kasprzak, J.M.; Tuszynska, I.; Magnus, M.; Szczepaniak, K.; Dawson, W.K.; Bujnicki, J.M. Modeling of Protein-RNA Complex Structures Using Computational Docking Methods. Methods Mol. Biol. 2016, 1414, 353–372. [Google Scholar] [CrossRef]
- Sun, L.Z.; Jiang, Y.; Zhou, Y.; Chen, S.J. RLDOCK: A New Method for Predicting RNA-Ligand Interactions. J. Chem. Theory Comput. 2020, 16, 7173–7183. [Google Scholar] [CrossRef]
- Kappel, K.; Das, R. Sampling Native-like Structures of RNA-Protein Complexes through Rosetta Folding and Docking. Structure 2019, 27, 140–151.e145. [Google Scholar] [CrossRef] [Green Version]
- Voss, N.R.; Gerstein, M. 3V: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555–W562. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Pisabarro, M.T. MSPocket: An orientation-independent algorithm for the detection of ligand binding pockets. Bioinformatics 2011, 27, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trosset, J.Y.; Vodovar, N. Structure-based target druggability assessment. Methods Mol. Biol. 2013, 986, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.G.; Sharp, K.A. Finding and characterizing tunnels in macromolecules with application to ion channels and pores. Biophys. J. 2009, 96, 632–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramo, T.; East, A.; Garzon, D.; Ulmschneider, M.B.; Bond, P.J. Efficient Characterization of Protein Cavities within Molecular Simulation Trajectories: Trj_cavity. J. Chem. Theory Comput. 2014, 10, 2151–2164. [Google Scholar] [CrossRef]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef] [Green Version]
- Laurent, B.; Chavent, M.; Cragnolini, T.; Dahl, A.C.; Pasquali, S.; Derreumaux, P.; Sansom, M.S.; Baaden, M. Epock: Rapid analysis of protein pocket dynamics. Bioinformatics 2015, 31, 1478–1480. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Wang, H.; Zeng, C.; Zhao, Y. RPocket: An intuitive database of RNA pocket topology information with RNA-ligand data resources. BMC Bioinformatics 2021, 22, 428–443. [Google Scholar] [CrossRef]
- Yang, R.; Liu, H.; Yang, L.; Zhou, T.; Li, X.; Zhao, Y. RPpocket: An RNA-Protein Intuitive Database with RNA Pocket Topology Resources. Int. J. Mol. Sci. 2022, 23, 6903. [Google Scholar] [CrossRef]
- Bhagavat, R.; Sankar, S.; Srinivasan, N.; Chandra, N. An Augmented Pocketome: Detection and Analysis of Small-Molecule Binding Pockets in Proteins of Known 3D Structure. Structure 2018, 26, 499–512.e492. [Google Scholar] [CrossRef] [Green Version]
- Donlic, A.; Swanson, E.G.; Chiu, L.Y.; Wicks, S.L.; Juru, A.U.; Cai, Z.; Kassam, K.; Laudeman, C.; Sanaba, B.G.; Sugarman, A.; et al. R-BIND 2.0: An Updated Database of Bioactive RNA-Targeting Small Molecules and Associated RNA Secondary Structures. ACS Chem. Biol. 2022, 17, 1556–1566. [Google Scholar] [CrossRef]
- Panei, F.P.; Torchet, R.; Menager, H.; Gkeka, P.; Bonomi, M. HARIBOSS: A curated database of RNA-small molecules structures to aid rational drug design. Bioinformatics 2022, 38, 4185–4193. [Google Scholar] [CrossRef]
- Kumar Mishra, S.; Kumar, A. NALDB: Nucleic acid ligand database for small molecules targeting nucleic acid. Database 2016, 2016, baw002. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Luo, H.; Teng, X.; Hao, X.; Yan, X.; Tang, Y.; Zhang, W.; Wang, Y.; Zhang, P.; Li, Y.; et al. NPInter v5.0: ncRNA interaction database in a new era. Nucleic Acids Res. 2022, 51, D232–D239. [Google Scholar] [CrossRef]
- Chagot, M.E.; Quinternet, M.; Rothe, B.; Charpentier, B.; Coutant, J.; Manival, X.; Lebars, I. The yeast C/D box snoRNA U14 adopts a "weak" K-turn like conformation recognized by the Snu13 core protein in solution. Biochimie 2019, 164, 70–82. [Google Scholar] [CrossRef]
- Cilley, C.D.; Williamson, J.R. Structural mimicry in the phage phi21 N peptide-boxB RNA complex. RNA 2003, 9, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Bahadur, R.P. A structural perspective of RNA recognition by intrinsically disordered proteins. Cell. Mol. Life Sci. 2016, 73, 4075–4084. [Google Scholar] [CrossRef]
- Varadi, M.; Zsolyomi, F.; Guharoy, M.; Tompa, P. Functional Advantages of Conserved Intrinsic Disorder in RNA-Binding Proteins. PLoS ONE 2015, 10, e0139731. [Google Scholar] [CrossRef]
- Cruz, M.A.; Frederick, T.E.; Mallimadugula, U.L.; Singh, S.; Vithani, N.; Zimmerman, M.I.; Porter, J.R.; Moeder, K.E.; Amarasinghe, G.K.; Bowman, G.R. A cryptic pocket in Ebola VP35 allosterically controls RNA binding. Nat. Commun. 2022, 13, 2269. [Google Scholar] [CrossRef]
- Schroeder, G.M.; Cavender, C.E.; Blau, M.E.; Jenkins, J.L.; Mathews, D.H.; Wedekind, J.E. A small RNA that cooperatively senses two stacked metabolites in one pocket for gene control. Nat. Commun. 2022, 13, 199. [Google Scholar] [CrossRef]
- Petushkov, I.; Pupov, D.; Bass, I.; Kulbachinskiy, A. Mutations in the CRE pocket of bacterial RNA polymerase affect multiple steps of transcription. Nucleic Acids Res. 2015, 43, 5798–5809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Ye, H.Q.; Deng, C.L.; Li, R.; Zhang, B.; Gong, P. A nucleobase-binding pocket in a viral RNA-dependent RNA polymerase contributes to elongation complex stability. Nucleic Acids Res. 2020, 48, 1392–1405. [Google Scholar] [CrossRef] [Green Version]
- De Zoysa, M.D.; Wu, G.; Katz, R.; Yu, Y.T. Guide-substrate base-pairing requirement for box H/ACA RNA-guided RNA pseudouridylation. RNA 2018, 24, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Li, H.; Xu, S.; Wang, J. Ligand Binding Mechanism and Its Relationship with Conformational Changes in Adenine Riboswitch. Int. J. Mol. Sci. 2020, 21, 1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kligun, E.; Mandel-Gutfreund, Y. Conformational readout of RNA by small ligands. RNA Biol. 2013, 10, 982–989. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef]
- Richards, F.M. Areas, volumes, packing and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176. [Google Scholar] [CrossRef]
- Greene, L.H.; Higman, V.A. Uncovering network systems within protein structures. J. Mol. Biol. 2003, 334, 781–791. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure Type | Number of Models | Complexes | Nucleotides | RNA Pockets |
|---|---|---|---|---|
| RNA | 5~10 | 47 | 2149 | 655 |
| 11~20 | 56 | 1894 | 1260 | |
| 21~30 | 12 | 405 | 322 | |
| 31~40 | 1 | 31 | 39 | |
| Total | 116 | 4479 | 2276 | |
| RNA–ligand | 5~10 | 12 | 432 | 155 |
| 11~20 | 5 | 183 | 119 | |
| 21~30 | 1 | 38 | 20 | |
| 31~40 | 1 | 27 | 58 | |
| Total | 19 | 680 | 352 | |
| RNA–protein | 5~10 | 6 | 215 | 59 |
| 11~20 | 16 | 493 | 332 | |
| 21~30 | 1 | 27 | 21 | |
| 31~40 | 2 | 60 | 114 | |
| Total | 25 | 795 | 526 |
| Testing Set | Number of Pocket Groups | Class | Binding Type | ||||
|---|---|---|---|---|---|---|---|
| Rigidity | Intermediate Flexibility | Flexibility | Non-Binding | Ligand-Binding | Protein-Binding | ||
| Small | 165 | 58 | 73 | 34 | 153 | 2 | 10 |
| Medium | 97 | 50 | 37 | 10 | 75 | 11 | 11 |
| Large | 35 | 22 | 9 | 4 | 31 | 2 | 2 |
| Total | 297 | 130 | 119 | 48 | 259 | 15 | 23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuo, C.; Zeng, C.; Yang, R.; Liu, H.; Zhao, Y. RPflex: A Coarse-Grained Network Model for RNA Pocket Flexibility Study. Int. J. Mol. Sci. 2023, 24, 5497. https://doi.org/10.3390/ijms24065497
Zhuo C, Zeng C, Yang R, Liu H, Zhao Y. RPflex: A Coarse-Grained Network Model for RNA Pocket Flexibility Study. International Journal of Molecular Sciences. 2023; 24(6):5497. https://doi.org/10.3390/ijms24065497
Chicago/Turabian StyleZhuo, Chen, Chengwei Zeng, Rui Yang, Haoquan Liu, and Yunjie Zhao. 2023. "RPflex: A Coarse-Grained Network Model for RNA Pocket Flexibility Study" International Journal of Molecular Sciences 24, no. 6: 5497. https://doi.org/10.3390/ijms24065497