
Modulation of Melatonin in Pain Behaviors Associated with Oxidative Stress and Neuroinflammation Responses in an Animal Model of Central Post-Stroke Pain

Abstract
:1. Introduction
2. Results
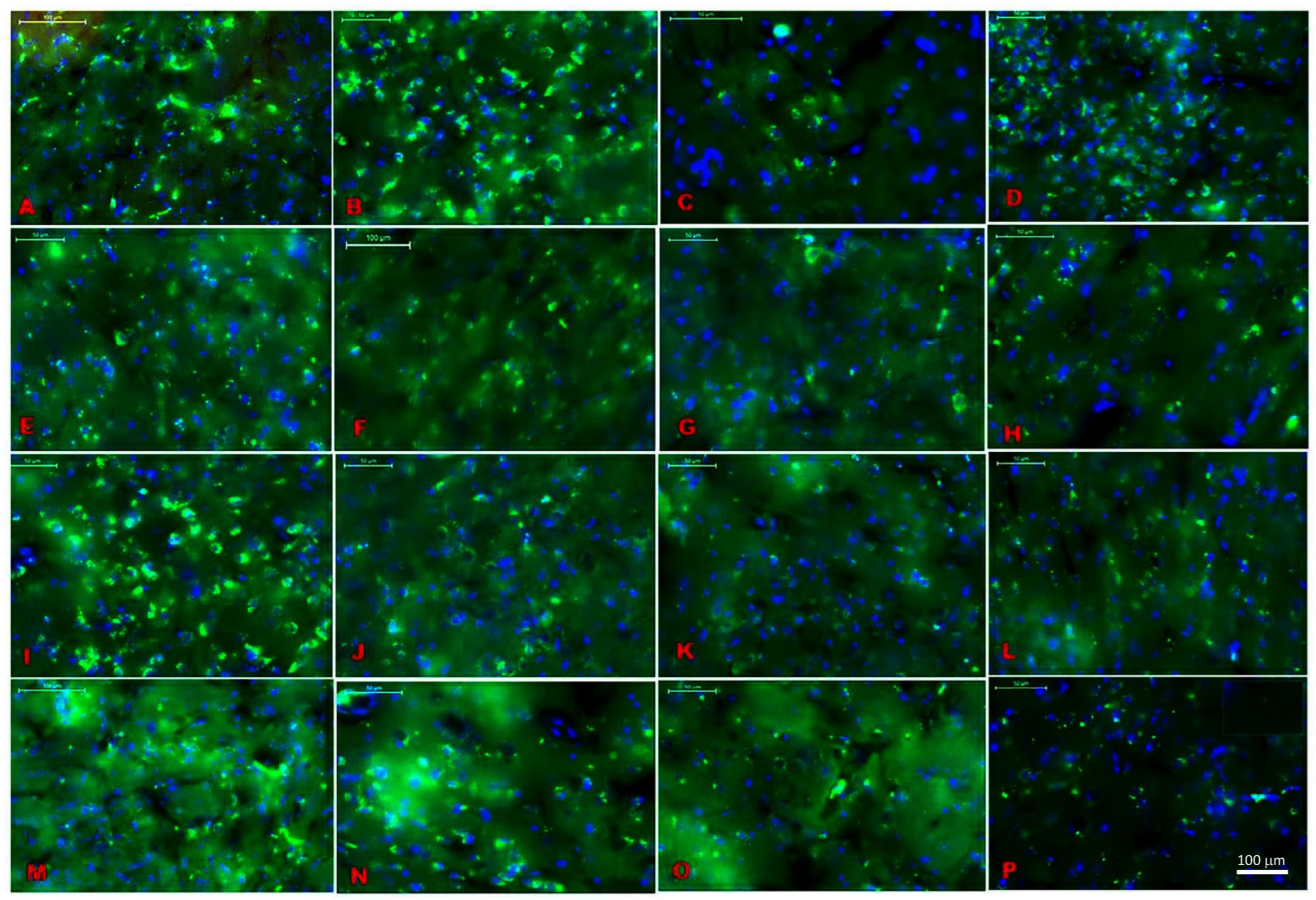
2.1. Location Assessments of Melatonin Receptors
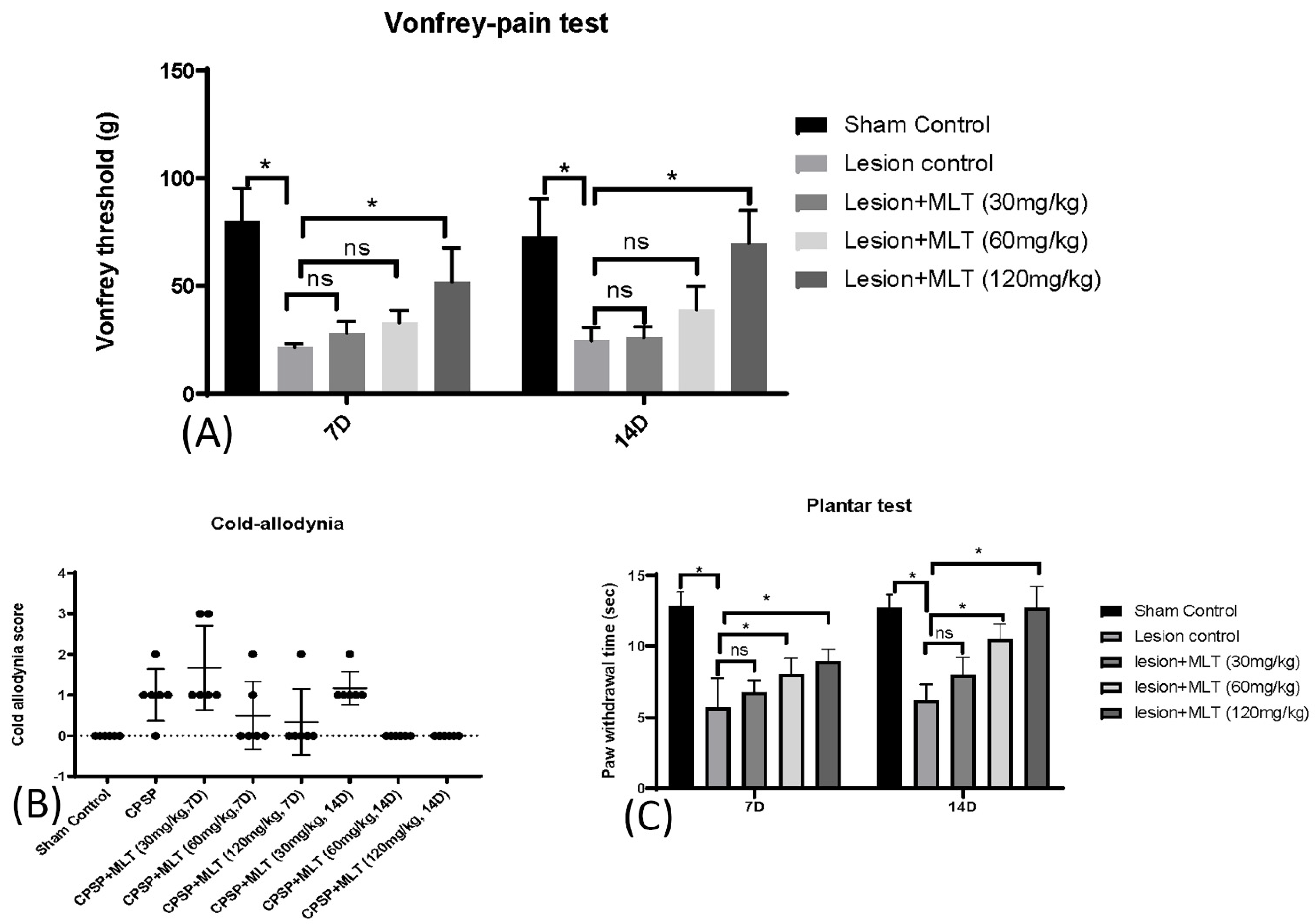
2.2. Induction of CPSP and Pain Behaviors
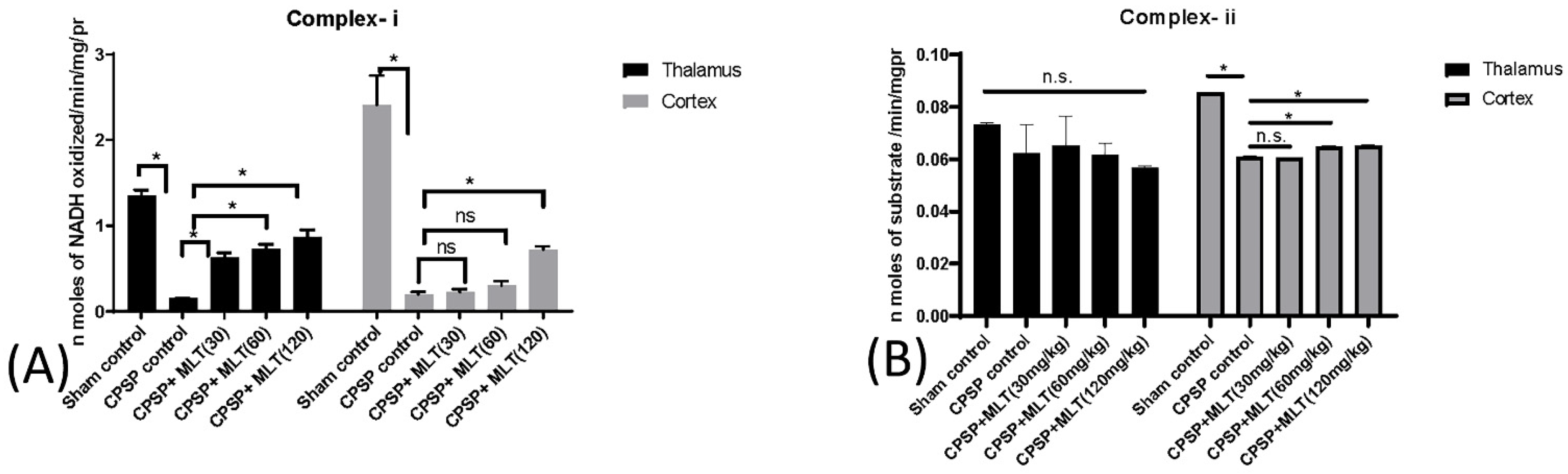
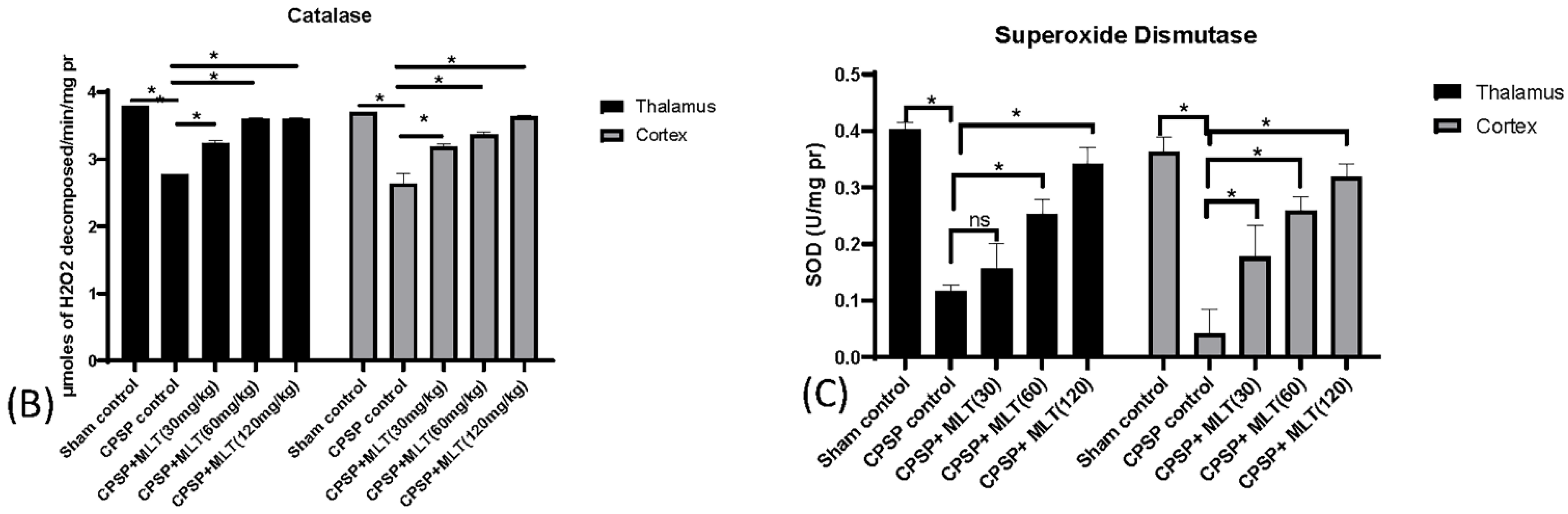
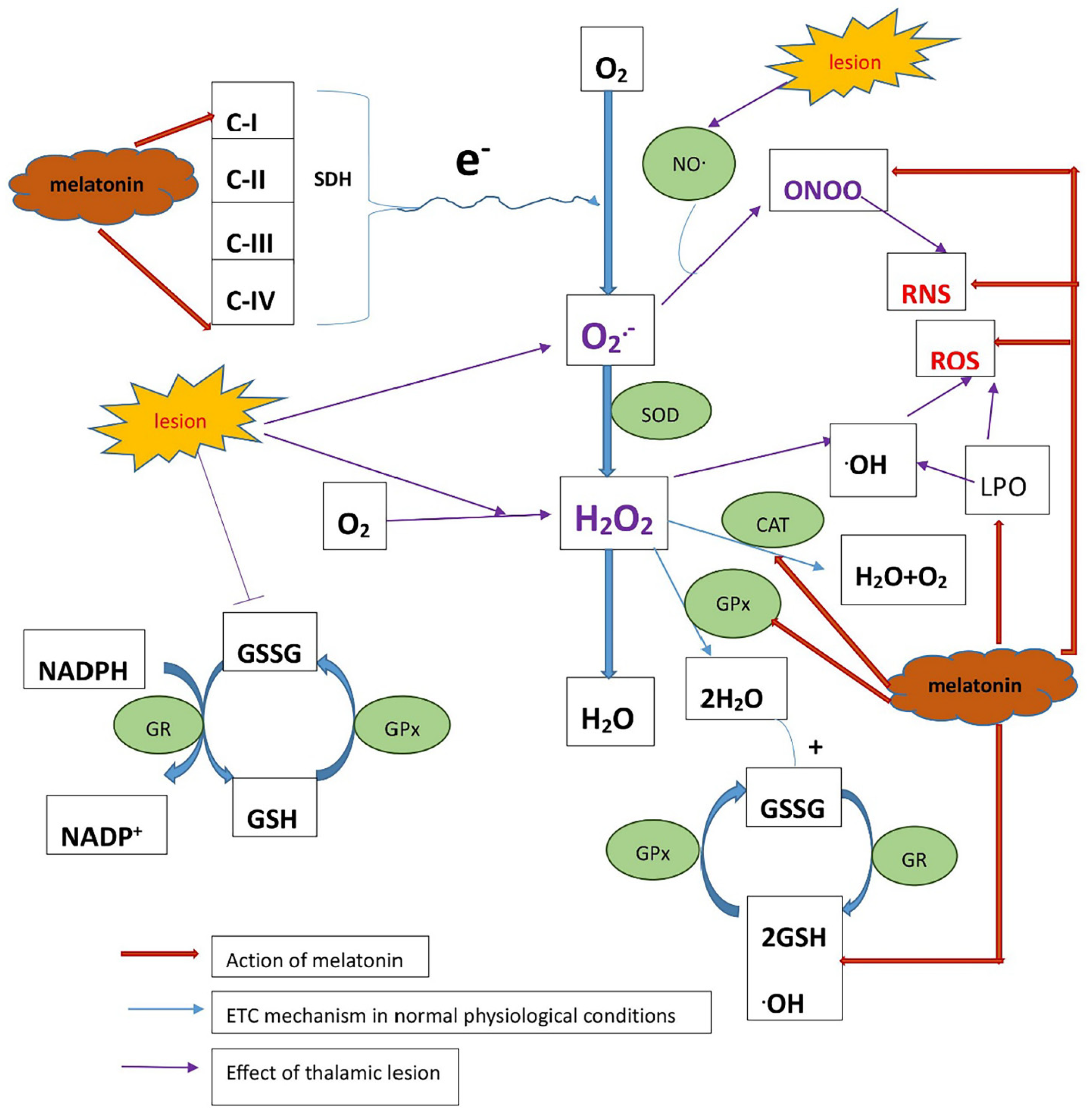
2.3. Determining Whether Melatonin Can Prevent Oxidative Stress Caused by Mitochondrial Dysfunction in CPSP Animal Model
2.3.1. Effect of Lesions on Electron Transport Chain Complexes and the Effect of Melatonin Treatment
2.3.2. Effect of Lesions on Electron Transport Chain Enzymes and the Effect of Melatonin Treatment
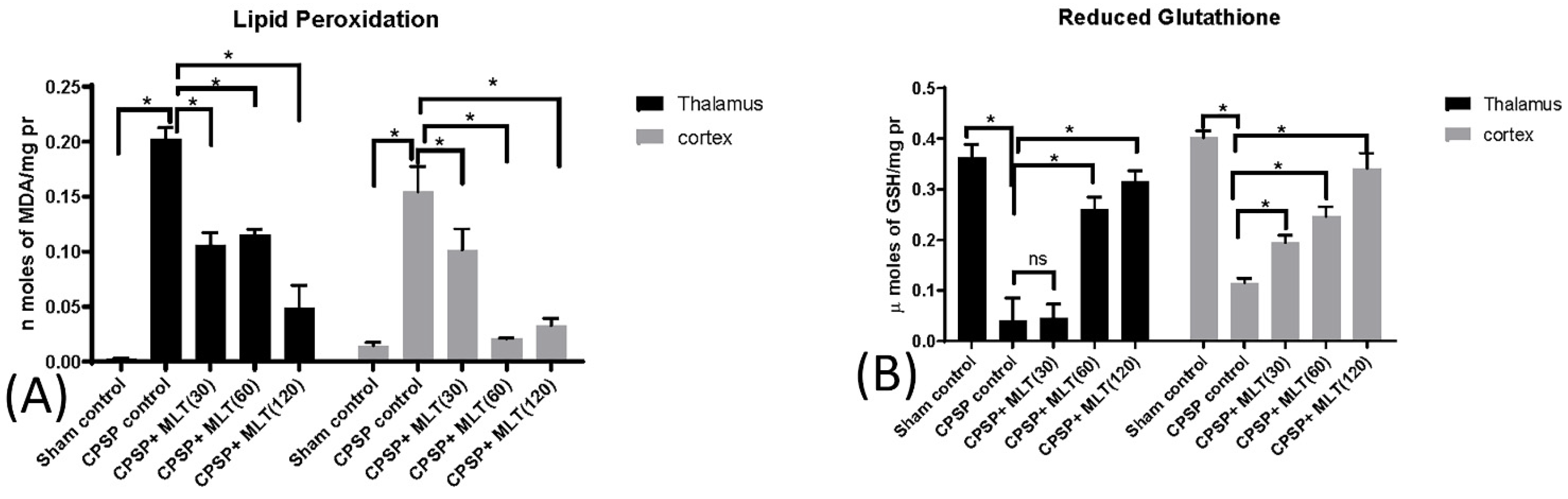
2.3.3. Effect of Lesion and Melatonin Treatments on Oxidative Stress
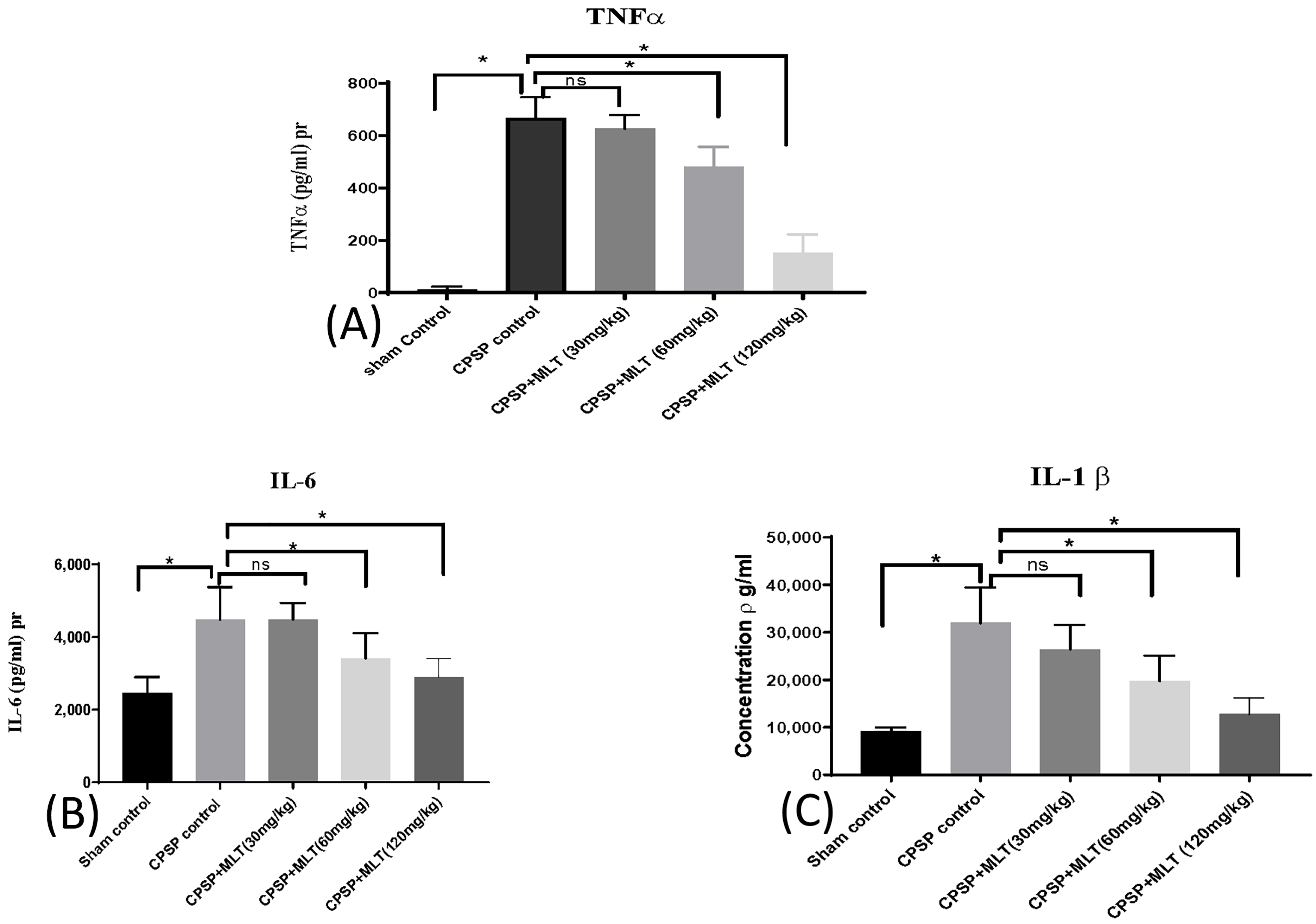
2.4. Effect of Melatonin on Cytokine Levels
3. Discussion
4. Materials and Methods
4.1. Animals and Drugs
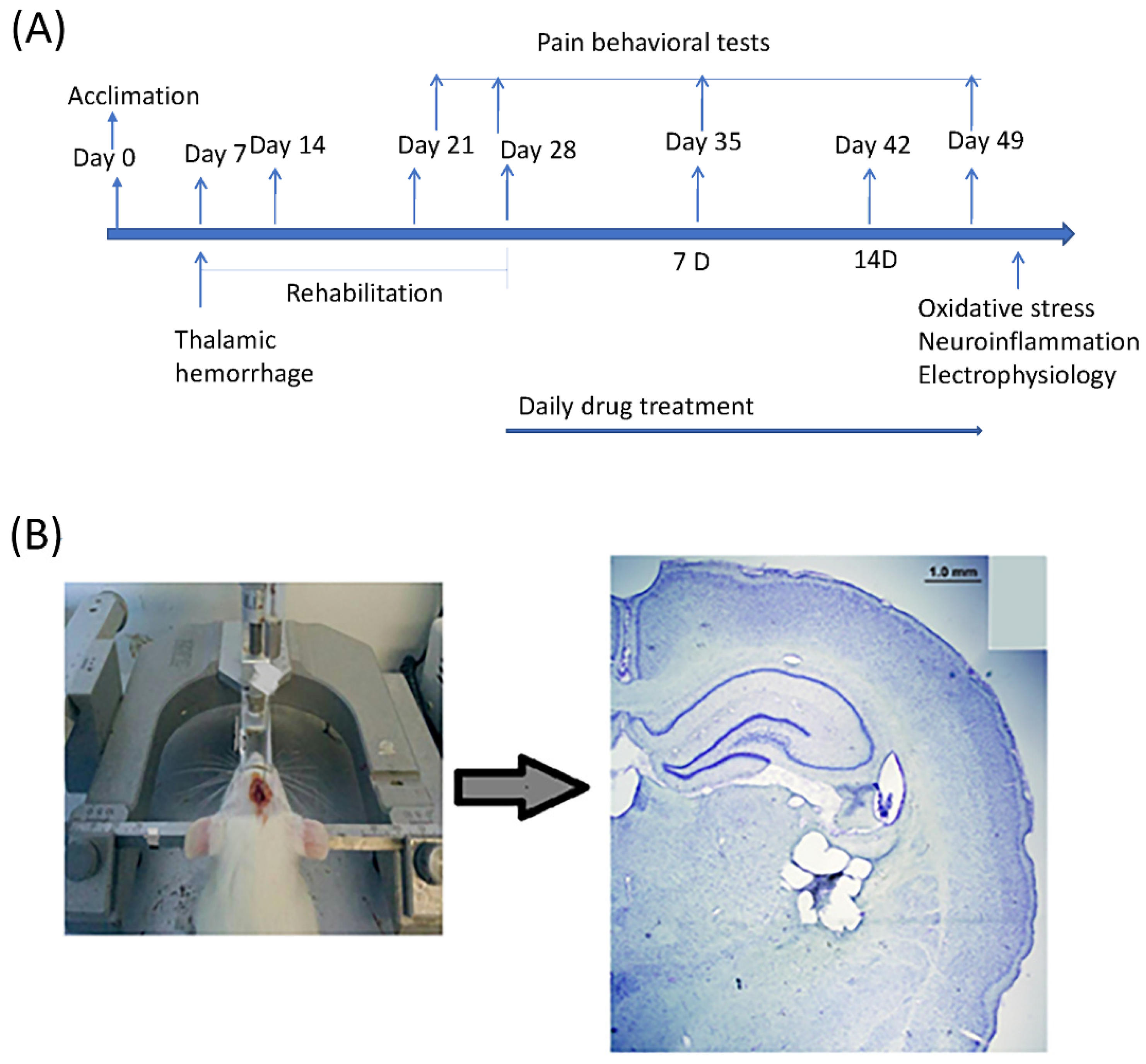
4.2. Experimental Procedure
4.3. CPSP Induction
4.4. MT1 Receptor Distribution in CNS: Using Immunohistochemistry
4.5. Pain Behavior Test
4.5.1. Von Frey
4.5.2. Plantar Test
4.5.3. Cold Allodynia
4.6. Experimental Procedures for Evaluating Oxidative Stress and Mitochondrial Function
4.6.1. Dissection and Homogenization
4.6.2. Isolation of Mitochondria
4.6.3. Protein Estimation
4.6.4. Estimation of Oxidative Stress Markers: Measurement of Lipid Peroxidation
4.6.5. Estimation of Oxidative Stress Markers: Estimation of Reduced Glutathione
4.6.6. Estimation of Mitochondrial Complexes and Enzymes
Estimation of Catalase (CAT) Activity
Determination of Glutathione Peroxidase (GPx) Activity
Estimation of Superoxide Dismutase (SOD) Activity
4.6.7. Estimation of Pro-Inflammatory Cytokines (IL-1β, IL-6, and TNF-α) Levels
4.7. Drug
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klit, H.; Finnerup, N.B.; Jensen, T.S. Central post-stroke pain: Clinical characteristics, pathophysiology, and management. Lancet Neurol. 2009, 8, 857–868. [Google Scholar] [CrossRef]
- Ntsiea, V. The prevalence and management of central post-stroke pain at a hospital in Zimbabwe. Malawi Med. J. 2020, 32, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Flatters, S.J. The contribution of mitochondria to sensory processing and pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 119–146. [Google Scholar]
- DiMauro, S.; Schon, E.A. Mitochondrial disorders in the nervous system. Annu. Rev. Neurosci. 2008, 31, 91–123. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Rezin, G.T.; Amboni, G.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Mitochondrial dysfunction and psychiatric disorders. Neurochem. Res. 2009, 34, 1021–1029. [Google Scholar] [CrossRef]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial dysfunction in Parkinson’s disease—Cause or consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef]
- O’Hanlon, M.E.; Tweedy, C.; Scialo, F.; Bass, R.; Sanz, A.; Smulders-Srinivasan, T.K. Mitochondrial electron transport chain defects modify Parkinson’s disease phenotypes in a Drosophila model. Neurobiol. Dis. 2022, 171, 105803. [Google Scholar] [CrossRef]
- Paul, K.N.; Saafir, T.B.; Tosini, G. The role of retinal photoreceptors in the regulation of circadian rhythms. Rev. Endocr. Metab. Disord. 2009, 10, 271–278. [Google Scholar] [CrossRef]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT1 and MT2 melatonin receptors: A therapeutic perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef]
- Cipolla-Neto, J.; Amaral, F.G.D. Melatonin as a hormone: New physiological and clinical insights. Endocr. Rev. 2018, 39, 990–1028. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Pain control by melatonin: Physiological and pharmacological effects. Exp. Ther. Med. 2016, 12, 1963–1968. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. Sci. World J. 2014, 2014, 586270. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.S.; Parvez, S.; Tabassum, H. Melatonin and ischemic stroke: Mechanistic roles and action. Adv. Pharmacol. Sci. 2015, 2015, 384750. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Canul, M.; Palazzo, E.; Dominguez-Lopez, S.; Luongo, L.; Lacoste, B.; Comai, S.; Angeloni, D.; Fraschini, F.; Boccella, S.; Spadoni, G. Selective melatonin MT2 receptor ligands relieve neuropathic pain through modulation of brainstem descending antinociceptive pathways. Pain 2015, 156, 305–317. [Google Scholar] [CrossRef]
- Castroviejo, D.A.; Lopez, L.C.; Escames, G.; Lopez, A.; Garcia, J.A.; Reiter, R.J. Melatonin-mitochondria interplay in health and disease. Curr. Top. Med. Chem. 2011, 11, 221–240. [Google Scholar] [CrossRef]
- Srinivasan, V.; Cardinali, D.P.; Srinivasan, U.S.; Kaur, C.; Brown, G.M.; Spence, D.W.; Hardeland, R.; Pandi-Perumal, S.R. Therapeutic potential of melatonin and its analogs in Parkinson’s disease: Focus on sleep and neuroprotection. Ther. Adv. Neurol. Disord. 2011, 4, 297–317. [Google Scholar] [CrossRef]
- Hardeland, R.; Pandi-Perumal, S. Melatonin, a potent agent in antioxidative defense: Actions as a natural food constituent, gastrointestinal factor, drug and prodrug. Nutr. Metab. 2005, 2, 22. [Google Scholar] [CrossRef]
- García, J.J.; López-Pingarrón, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; García-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramírez, J.M.; Bernal-Pérez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Gbahou, F.; Jockers, R. 2-[125I] Iodomelatonin and [3H] Melatonin Binding Assays for Melatonin Receptors. In Melatonin; Springer: Berlin/Heidelberg, Germany, 2022; pp. 141–149. [Google Scholar]
- Persengiev, S.P. 2-(125I) iodomelatonin binding sites in rat adrenals: Pharmacological characteristics and subcellular distribution. Life Sci. 1992, 51, 647–651. [Google Scholar] [CrossRef]
- Slominski, R.M.; Reiter, R.J.; Schlabritz-Loutsevitch, N.; Ostrom, R.S.; Slominski, A.T. Melatonin membrane receptors in peripheral tissues: Distribution and functions. Mol. Cell. Endocrinol. 2012, 351, 152–166. [Google Scholar] [CrossRef]
- Kaur, T.; Shih, H.-C.; Huang, A.C.W.; Shyu, B.-C. Modulation of melatonin to the thalamic lesion-induced pain and comorbid sleep disturbance in the animal model of the central post-stroke hemorrhage. Mol. Pain 2022, 18, 17448069221127180. [Google Scholar] [CrossRef]
- Jeelani, R.; Maitra, D.; Chatzicharalampous, C.; Najeemuddin, S.; Morris, R.T.; Abu-Soud, H.M. Melatonin prevents hypochlorous acid-mediated cyanocobalamin destruction and cyanogen chloride generation. J. Pineal Res. 2018, 64, e12463. [Google Scholar] [CrossRef]
- Srinivasan, V.; Spence, D.W.; Pandi-Perumal, S.R.; Brown, G.M.; Cardinali, D.P. Melatonin in mitochondrial dysfunction and related disorders. Int. J. Alzheimer’s Dis. 2011, 2011, 326320. [Google Scholar] [CrossRef]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK p38 regulates transcriptional activity of NF-κB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef]
- Kuan, Y.-H.; Shih, H.-C.; Tang, S.-C.; Jeng, J.-S.; Shyu, B.-C. Targeting P2X7 receptor for the treatment of central post-stroke pain in a rodent model. Neurobiol. Dis. 2015, 78, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Flatters, S.J.; Bennett, G.J. Ethosuximide reverses paclitaxel-and vincristine-induced painful peripheral neuropathy. Pain 2004, 109, 150–161. [Google Scholar] [CrossRef]
- Aziz, C.B.A.; Ahmad, A.H. The role of the thalamus in modulating pain. Malays. J. Med. Sci. MJMS 2006, 13, 11–18. [Google Scholar]
- Xie, Y.F.; Huo, F.Q.; Tang, J.S. Cerebral cortex modulation of pain. Acta Pharmacol. Sin. 2009, 30, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Gómez, C.; Sánchez-Pino, M.-J.; González, H.; Bández, M.J.; Boveris, A.D.; Boveris, A. Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1392–R1399. [Google Scholar] [CrossRef]
- Lowry, O.H. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Wills, E. Mechanisms of lipid peroxide formation in animal tissues. Biochem. J. 1966, 99, 667. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Birt, D.; Schnell, R. Direct enzymatic assay for reduced and oxidized glutathione. J. Pharmacol. Methods 1984, 12, 191–194. [Google Scholar] [CrossRef]
- King, T.E.; Howard, R.L. [52] Preparations and Properties of Soluble NADH Dehydrogenases from Cardiac Muscle. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1967; Volume 10, pp. 275–294. [Google Scholar]
- Almeida, A.; Allen, K.L.; Bates, T.E.; Clark, J.B. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J. Neurochem. 1995, 65, 1698–1703. [Google Scholar] [CrossRef]
- Surin, A.; Sharipov, R.; Krasil’Nikova, I.; Boyarkin, D.; Lisina, O.Y.; Gorbacheva, L.; Avetisyan, A.; Pinelis, V. Disruption of functional activity of mitochondria during MTT assay of viability of cultured neurons. Biochemistry 2017, 82, 737–749. [Google Scholar] [CrossRef]
- Sottocasa, G.L.; Kuylenstierna, B.; Ernster, L.; Bergstrand, A. An electron-transport system associated with the outer membrane of liver mitochondria: A biochemical and morphological study. J. Cell Biol. 1967, 32, 415–438. [Google Scholar] [CrossRef]
- Luck, H. Catalase estimation. In Method of Enzymatic Analysis; Academic Press: London, UK, 1971. [Google Scholar]
- Kono, Y. Generation of superoxide radical during autoxidation of hydroxylamine and an assay for superoxide dismutase. Arch. Biochem. Biophys. 1978, 186, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-X.; Zhu, C.-B.; Xu, S.-F.; Cao, X.-D.; Wu, G.-C. The analgesic effects of peripheral and central administration of melatonin in rats. Eur. J. Pharmacol. 2000, 403, 49–53. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region of Interest | Annotation Area | Relative Mask Area | Total Count | Receptor Density |
|---|---|---|---|---|
| S1 | 7.67 mm2 | 0.12% | 3170 | 413.1 mm2 |
| VM | 0.47 mm2 | 0.1% | 211 | 449.45 mm2 |
| RT | 0.34 mm2 | 0.23% | 265 | 768.52 mm2 |
| Hippocampus | 5.50 mm2 | 1.14% | 4648 | 844.86 mm2 |
| MEPD/MEPV | 0.42 mm2 | 0.2% | 415 | 996.89 mm2 |
| VPM/VPL | 2.26 mm2 | 0.88% | 3274 | 1450.59 mm2 |
| LH (lateral hypothalamus) | 0.68 mm2 | 0.44% | 592 | 867.58 mm2 |
| Vmhyp nu (ventromedial hypothalamus) | 2.12 mm2 | 0.09% | 579 | 273.11 mm2 |
| AUD | 4.11 mm2 | 0.33% | 3428 | 833.96 mm2 |
| PH (posterior hypothalamus) | 0.26 mm2 | 0.27% | 163 | 635.45 mm2 |
| BLA/BMP | 0.73 mm2 | 0.08% | 223 | 303.86 mm2 |
| S2 | 1.01 mm2 | 0.23% | 751 | 744.1 mm2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, T.; Huang, A.C.-W.; Shyu, B.-C. Modulation of Melatonin in Pain Behaviors Associated with Oxidative Stress and Neuroinflammation Responses in an Animal Model of Central Post-Stroke Pain. Int. J. Mol. Sci. 2023, 24, 5413. https://doi.org/10.3390/ijms24065413
Kaur T, Huang AC-W, Shyu B-C. Modulation of Melatonin in Pain Behaviors Associated with Oxidative Stress and Neuroinflammation Responses in an Animal Model of Central Post-Stroke Pain. International Journal of Molecular Sciences. 2023; 24(6):5413. https://doi.org/10.3390/ijms24065413
Chicago/Turabian StyleKaur, Tavleen, Andrew Chih-Wei Huang, and Bai-Chuang Shyu. 2023. "Modulation of Melatonin in Pain Behaviors Associated with Oxidative Stress and Neuroinflammation Responses in an Animal Model of Central Post-Stroke Pain" International Journal of Molecular Sciences 24, no. 6: 5413. https://doi.org/10.3390/ijms24065413