
Developmental Toxicity Studies: The Path towards Humanized 3D Stem Cell-Based Models
 ,
,  and
and
Abstract
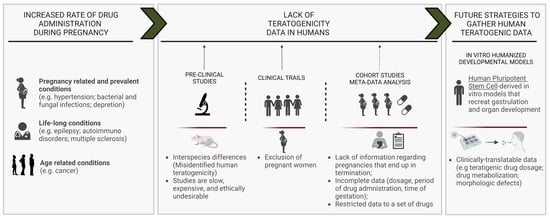
:
1. Introduction
2. Animal Models in Developmental Toxicity Assessment
3. In Vitro Animal-Based Models to Assess Teratogenicity
4. In Vitro Humanized Models to Assess Developmental Toxicity
5. Multilineage Developmental Toxicity Studies—Use of HPSC-Derived EBs and Gastruloids
6. Cardiac Developmental Toxicity Studies—Use of HPSC-Derived 2D and 3D Models
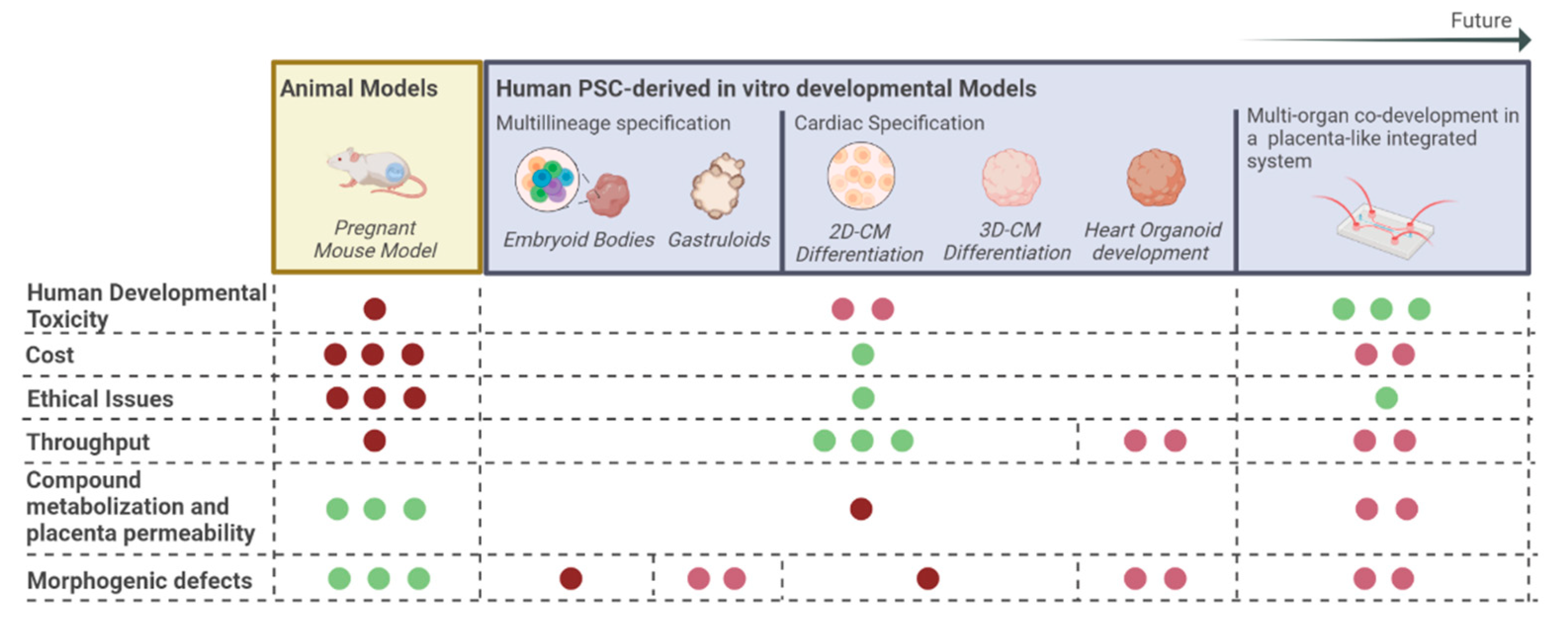
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- EMA. ICH S5 (R3) Guideline on Reproductive Toxicology: Detection of Toxicity to Reproduction for Human Pharmaceuticals Step 5; European Medicines Agency, Committee for Medicinal Products for Human Use: Amsterdam, The Netherlands, 2020; Volume 5. [Google Scholar]
- Adam, M.P.; Polifka, J.E.; Friedman, J.M. Evolving knowledge of the teratogenicity of medications in human pregnancy. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Mazer-Amirshahi, M.; Samiee-Zafarghandy, S.; Gray, G.; van den Anker, J.N. Trends in pregnancy labeling and data quality for US-approved pharmaceuticals. Am. J. Obstet. Gynecol. 2014, 211, 690.e1–690.e11. [Google Scholar] [CrossRef] [PubMed]
- Shields, K.E.; Lyerly, A.D. Exclusion of Pregnant Women From Industry-Sponsored Clinical Trials. Obstet. Gynecol. 2013, 122, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- McCormack, S.A.; Best, B.M. Obstetric pharmacokinetic dosing studies are urgently needed. Front. Pediatr. 2014, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Ayad, M. Epidemiology of Medications Use in Pregnancy Martina. Semin. Perinatol. 2018, 39, 508–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finer, L.B.; Zolna, M.R. Shifts in Intended and Unintended Pregnancies in the United States, 2001–2008. Am. J. Public Health 2014, 104, S43–S48. [Google Scholar] [CrossRef]
- Platzbecker, K.; Wentzell, N.; Kollhorst, B.; Haug, U. Fingolimod, teriflunomide and cladribine for the treatment of multiple sclerosis in women of childbearing age: Description of drug utilization and exposed pregnancies in Germany. Mult. Scler. Relat. Disord. 2022, 67, 104184. [Google Scholar] [CrossRef]
- Leke, A.Z.; Dolk, H.; Loane, M.; Casson, K.; Nelen, V.; Barišić, I.; Garne, E.; Rissman, A.; O’Mahony, M.; Neville, A.J.; et al. Macrolide and lincosamide antibiotic exposure in the first trimester of pregnancy and risk of congenital anomaly: A European case-control study. Reprod. Toxicol. 2021, 100, 101–108. [Google Scholar] [CrossRef]
- Jawaid, S.; Strainic, J.P.; Kim, J.; Ford, M.R.; Thrane, L.; Karunamuni, G.H.; Sheehan, M.M.; Chowdhury, A.; Gillespie, C.A.; Rollins, A.M.; et al. Glutathione Protects the Developing Heart from Defects and Global DNA Hypomethylation Induced by Prenatal Alcohol Exposure. Alcohol. Clin. Exp. Res. 2021, 45, 69–78. [Google Scholar] [CrossRef]
- Bookstaver, P.B.; Bland, C.M.; Griffin, B.; Stover, K.R.; Eiland, L.S.; McLaughlin, M. A Review of Antibiotic Use in Pregnancy. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2015, 35, 1052–1062. [Google Scholar] [CrossRef]
- Anderson, K.N.; Lind, J.N.; Simeone, R.M.; Bobo, W.V.; Mitchell, A.A.; Riehle-Colarusso, T.; Polen, K.N.; Reefhuis, J. Maternal Use of Specific Antidepressant Medications During Early Pregnancy and the Risk of Selected Birth Defects. JAMA Psychiatry 2020, 77, 1246. [Google Scholar] [CrossRef] [PubMed]
- Bérard, A.; Iessa, N.; Chaabane, S.; Muanda, F.T.; Boukhris, T.; Zhao, J.P. The risk of major cardiac malformations associated with paroxetine use during the first trimester of pregnancy: A systematic review and meta-analysis. Br. J. Clin. Pharmacol. 2016, 81, 589–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Qiu, J.; Zhang, S.; Zhang, L. Fetal Congenital Cardiac and Vascular Disorders Associated with Sertraline Treatment during Pregnancy: Analysis of FAERS Data. BioMed Res. Int. 2022, 2022, 9914931. [Google Scholar] [CrossRef]
- Kolding, L.; Ehrenstein, V.; Pedersen, L.; Sandager, P.; Petersen, O.; Uldbjerg, N.; Pedersen, L. Antidepressant use in pregnancy and severe cardiac malformations: Danish register-based study. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Koutras, A.; Ntounis, T.; Fasoulakis, Z.; Papalios, T.; Pittokopitou, S.; Prokopakis, I.; Syllaios, A.; Valsamaki, A.; Chionis, A.; Symeonidis, P.; et al. Cancer Treatment and Immunotherapy during Pregnancy. Pharmaceutics 2022, 14, 2080. [Google Scholar] [CrossRef]
- Maggen, C.; Wolters, V.E.R.A.; Cardonick, E.; Fumagalli, M.; Halaska, M.J.; Lok, C.A.R.; de Haan, J.; Van Tornout, K.; Van Calsteren, K.; Amant, F. Pregnancy and Cancer: The INCIP Project. Curr. Oncol. Rep. 2020, 22, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomson, T.; Battino, D. Teratogenic Effects of Antiepileptic Medications. Neurol. Clin. 2009, 27, 993–1002. [Google Scholar] [CrossRef]
- Fried, S.; Kozer, E.; Nulman, I.; Einarson, T.R.; Koren, G. Malformation Rates in Children of Women with Untreated Epilepsy. Drug Saf. 2004, 27, 197–202. [Google Scholar] [CrossRef]
- Duley, L. The Global Impact of Pre-eclampsia and Eclampsia. Semin. Perinatol. 2009, 33, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.; McMullen, H.; Beqaj, H.; Kalfa, D. Environmental Exposures and Congenital Heart Disease. Pediatrics 2022, 149, e2021052151. [Google Scholar] [CrossRef]
- Pruss-Ustun, A.C.C. Preventing Disease through Healthy Environments: Towards an Estimate of the Environmental Burden of Diseasee. Available online: https://apps.who.int/iris/handle/10665/43457 (accessed on 30 January 2023).
- Mahler, G.J.; Butcher, J.T. Cardiac developmental toxicity. Birth Defects Res. Part C Embryo Today Rev. 2011, 93, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, A.; Samtani, R.; Dhanantwari, P.; Lee, E.; Yamada, S.; Shiota, K.; Donofrio, M.T.; Leatherbury, L.; Lo, C.W. A detailed comparison of mouse and human cardiac development. Pediatr. Res. 2014, 76, 500–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheradvar, A.; Zareian, R.; Kawauchi, S.; Goodwin, R.L.; Rugonyi, S. Animal models for heart valve research and development. Drug Discov. Today Dis. Model. 2017, 24, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Meng, Z.; Ruan, H.; Yin, W.; Xu, Y.; Zhang, T. Heart Development and Regeneration in Non-mammalian Model Organisms. Front. Cell Dev. Biol. 2020, 8, 595488. [Google Scholar] [CrossRef]
- Zakaria, Z.Z.; Benslimane, F.M.; Nasrallah, G.K.; Shurbaji, S.; Younes, N.N.; Mraiche, F.; Da’as, S.I.; Yalcin, H.C. Using Zebrafish for Investigating the Molecular Mechanisms of Drug-Induced Cardiotoxicity. Biomed Res. Int. 2018, 2018, 1642684. [Google Scholar] [CrossRef] [Green Version]
- Scialli, A.R. The challenge of reproductive and developmental toxicology under REACH. Regul. Toxicol. Pharmacol. 2008, 51, 244–250. [Google Scholar] [CrossRef]
- Augustine-Rauch, K.; Zhang, C.X.; Panzica-Kelly, J.M. In vitro developmental toxicology assays: A review of the state of the science of rodent and zebrafish whole embryo culture and embryonic stem cell assays. Birth Defects Res. Part C Embryo Today Rev. 2010, 90, 87–98. [Google Scholar] [CrossRef]
- Mohammed, O.J.; Pratten, M.K. Micromass Methods for the Evaluation of Developmental Toxicants. In Developmental Toxicology: Methods and Protocols; Humana: New York, NY, USA, 2019; pp. 49–72. [Google Scholar]
- Lauschke, K.; Rosenmai, A.K.; Meiser, I.; Neubauer, J.C.; Schmidt, K.; Rasmussen, M.A.; Holst, B.; Taxvig, C.; Emnéus, J.K.; Vinggaard, A.M. A novel human pluripotent stem cell-based assay to predict developmental toxicity. Arch. Toxicol. 2020, 94, 3831–3846. [Google Scholar] [CrossRef]
- Daston, G.P.; Knudsen, T.B. Fundamental concepts, current regulatory design and interpretation. Compr. Toxicol. Elsevier 2010, 12, 3–9. [Google Scholar]
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide–identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Flamier, A.; Singh, S.; Rasmussen, T.P. A standardized human embryoid body platform for the detection and analysis of teratogens. PLoS ONE 2017, 12, e0171101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, V.; Perumalinivasan, S.; Henry, M.; Rotshteyn, T.; Hescheler, J.; Rahnenführer, J.; Grinberg, M.; Meisig, J.; Blüthgen, N.; Waldmann, T.; et al. Comparison of a teratogenic transcriptome-based predictive test based on human embryonic versus inducible pluripotent stem cells. Stem Cell Res. Ther. 2016, 7, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konala, V.B.R.; Nandakumar, S.; Surendran, H.; Datar, S.; Bhonde, R.; Pal, R. Neuronal and cardiac toxicity of pharmacological compounds identified through transcriptomic analysis of human pluripotent stem cell-derived embryoid bodies. Toxicol. Appl. Pharmacol. 2021, 433, 115792. [Google Scholar] [CrossRef]
- Meganathan, K.; Jagtap, S.; Wagh, V.; Winkler, J.; Gaspar, J.A.; Hildebrand, D.; Trusch, M.; Lehmann, K.; Hescheler, J.; Schlüter, H.; et al. Identification of Thalidomide-Specific Transcriptomics and Proteomics Signatures during Differentiation of Human Embryonic Stem Cells. PLoS ONE 2012, 7, e44228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaklin, M.; Zhang, J.D.; Barrow, P.; Ebeling, M.; Clemann, N.; Leist, M.; Kustermann, S. Focus on germ-layer markers: A human stem cell-based model for in vitro teratogenicity testing. Reprod. Toxicol. 2020, 98, 286–298. [Google Scholar] [CrossRef]
- Guo, H.; Tian, L.; Zhang, J.Z.; Kitani, T.; Paik, D.T.; Lee, W.H.; Wu, J.C. Single-Cell RNA Sequencing of Human Embryonic Stem Cell Differentiation Delineates Adverse Effects of Nicotine on Embryonic Development. Stem Cell Rep. 2019, 12, 772–786. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Chen, Y.; Luz, A.; Hu, G.; Tokar, E.J. Cardiac Development in the Presence of Cadmium: An in Vitro Study Using Human Embryonic Stem Cells and Cardiac Organoids. Environ. Health Perspect. 2022, 130, 117002. [Google Scholar] [CrossRef]
- Mantziou, V.; Baillie-Benson, P.; Jaklin, M.; Kustermann, S.; Arias, A.M.; Moris, N. In vitro teratogenicity testing using a 3D, embryo-like gastruloid system. Reprod. Toxicol. 2021, 105, 72–90. [Google Scholar] [CrossRef]
- Kirkwood-Johnson, L.; Marikawa, Y. Developmental toxicity of remdesivir, an anti-COVID-19 drug, is implicated by in vitro assays using morphogenetic embryoid bodies of mouse and human pluripotent stem cells. Birth Defects Res. 2022, 115, 224–239. [Google Scholar] [CrossRef]
- Kameoka, S.; Babiarz, J.; Kolaja, K.; Chiao, E. A High-Throughput Screen for Teratogens Using Human Pluripotent Stem Cells. Toxicol. Sci. 2014, 137, 76–90. [Google Scholar] [CrossRef]
- Xing, J.; Toh, Y.C.; Xu, S.; Yu, H. A method for human teratogen detection by geometrically confined cell differentiation and migration. Sci. Rep. 2015, 5, 10038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Cao, Y.; Yu, Y.; Li, H.; Song, Z.; Yu, H. In Vitro Micropatterned Human Pluripotent Stem Cell Test (μP-hPST) for Morphometric-Based Teratogen Screening. Sci. Rep. 2017, 7, 8491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Van Bortle, K.; Zhang, Y.; Zhao, M.T.; Zhang, J.Z.; Geller, B.S.; Gruber, J.J.; Jiang, C.; Wu, J.C.; Snyder, M.P. Disruption of mesoderm formation during cardiac differentiation due to developmental exposure to 13-cis-retinoic acid. Sci. Rep. 2018, 8, 12960. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Han, Z.; Zhang, B.; Yu, Y.; Xu, Z.; Ma, W.; Ding, F.; Zhang, L.; Yu, M.; Liu, S.; et al. Arsenic trioxide blocked proliferation and cardiomyocyte differentiation of human induced pluripotent stem cells: Implication in cardiac developmental toxicity. Toxicol. Lett. 2019, 309, 51–58. [Google Scholar] [CrossRef]
- Fu, H.; Wang, L.; Wang, J.; Bennett, B.D.; Li, J.; Zhao, B.; Hu, G.; Hospital, F.; Medical, P.U.; Cell, S.; et al. HHS Public Access. Sci. Total Environ. 2020, 651, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Bao, Z.; Yu, Y.; Han, Z.; Yu, Y.; Xu, Z.; Ma, W.; Yuan, Y.; Zhang, L.; Xu, Y.; et al. Inhibition of cardiomyocyte differentiation of human induced pluripotent stem cells by Ribavirin: Implication for its cardiac developmental toxicity. Toxicology 2020, 435, 152422. [Google Scholar] [CrossRef]
- Jamalpoor, A.; Hartvelt, S.; Dimopoulou, M.; Zwetsloot, T.; Brandsma, I.; Racz, P.I.; Osterlund, T.; Hendriks, G. A novel human stem cell-based biomarker assay for in vitro assessment of developmental toxicity. Birth Defects Res. 2022, 114, 1210–1228. [Google Scholar] [CrossRef]
- Galanjuk, S.; Zühr, E.; Dönmez, A.; Bartsch, D.; Kurian, L.; Tigges, J.; Fritsche, E. The Human Induced Pluripotent Stem Cell Test as an Alternative Method for Embryotoxicity Testing. Int. J. Mol. Sci. 2022, 23, 3295. [Google Scholar] [CrossRef]
- Lauschke, K.; Treschow, A.F.; Rasmussen, M.A.; Davidsen, N.; Holst, B.; Emnéus, J.; Taxvig, C.; Vinggaard, A.M. Creating a human-induced pluripotent stem cell-based NKX2.5 reporter gene assay for developmental toxicity testing. Arch. Toxicol. 2021, 95, 1659–1670. [Google Scholar] [CrossRef]
- Schmidt, C.; Deyett, A.; Ilmer, T.; Caballero, A.T.; Haendeler, S.; Pimpale, L.; Netzer, M.A.; Ginistrelli, L.C.; Cirigliano, M.; Mancheno, E.J.; et al. Multi-chamber cardioids unravel human heart development and cardiac defects. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hoang, P.; Kowalczewski, A.; Sun, S.; Winston, T.S.; Archilla, A.M.; Lemus, S.M.; Ercan-Sencicek, A.G.; Gupta, A.R.; Liu, W.; Kontaridis, M.I.; et al. Engineering spatial-organized cardiac organoids for developmental toxicity testing. Stem Cell Rep. 2021, 16, 1228–1244. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yin, F.; Wang, H.; Wang, L.; Yuan, J.; Qin, J. Placental Barrier-on-a-Chip: Modeling Placental Inflammatory Responses to Bacterial Infection. ACS Biomater. Sci. Eng. 2018, 4, 3356–3363. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Romero, R.; Han, Y.M.; Kim, H.C.; Kim, C.J.; Hong, J.S.; Huh, D. Placenta-on-A-chip: A novel platform to study the biology of the human placenta. J. Matern. Neonatal Med. 2016, 29, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Blundell, C.; Yi, Y.-S.; Ma, L.; Tess, E.R.; Farrell, M.J.; Georgescu, A.; Aleksunes, L.M.; Huh, D. Placental Drug Transport-on-a-Chip: A Microengineered In Vitro Model of Transporter-Mediated Drug Efflux in the Human Placental Barrier. Adv. Healthc. Mater. 2018, 7, 1700786. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Zhu, Y.; Zhang, M.; Yu, H.; Chen, W.; Qin, J. A 3D human placenta-on-a-chip model to probe nanoparticle exposure at the placental barrier. Toxicol. Vitr. 2019, 54, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Pemathilaka, R.L.; Caplin, J.D.; Aykar, S.S.; Montazami, R.; Hashemi, N.N. Placenta-on-a-Chip: In Vitro Study of Caffeine Transport across Placental Barrier Using Liquid Chromatography Mass Spectrometry. Glob. Chall. 2019, 3, 1800112. [Google Scholar] [CrossRef] [Green Version]
- Cinkornpumin, J.K.; Kwon, S.Y.; Guo, Y.; Hossain, I.; Sirois, J.; Russett, C.S.; Tseng, H.W.; Okae, H.; Arima, T.; Duchaine, T.F.; et al. Naive Human Embryonic Stem Cells Can Give Rise to Cells with a Trophoblast-like Transcriptome and Methylome. Stem Cell Rep. 2020, 15, 198–213. [Google Scholar] [CrossRef]
- Dong, C.; Beltcheva, M.; Gontarz, P.; Zhang, B.; Popli, P.; Fischer, L.A.; Khan, S.A.; Park, K.M.; Yoon, E.J.; Xing, X.; et al. Derivation of trophoblast stem cells from naïve human pluripotent stem cells. Elife 2020, 9, e52504. [Google Scholar] [CrossRef]
- Soininen, S.K.; Repo, J.K.; Karttunen, V.; Auriola, S.; Vähäkangas, K.H.; Ruponen, M. Human placental cell and tissue uptake of doxorubicin and its liposomal formulations. Toxicol. Lett. 2015, 239, 108–114. [Google Scholar] [CrossRef]
- Tenreiro, M.F.; Branco, M.A.; Cotovio, J.P.; Cabral, J.M.S.; Fernandes, T.G.; Diogo, M.M. Advancing organoid design through co-emergence, assembly, and bioengineering. Trends Biotechnol. 2023, 1–16. [Google Scholar] [CrossRef]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.J.; Krotenberg Garcia, A.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879.e11. [Google Scholar] [CrossRef]
- Giacomelli, E.; Bellin, M.; Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Orlova, V.V.; Mummery, C.L. Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development 2017, 144, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Branco, M.A.; Cotovio, J.P.; Rodrigues, C.A.V.; Vaz, S.H.; Fernandes, T.G.; Moreira, L.M.; Cabral, J.M.S.; Diogo, M.M. Transcriptomic analysis of 3D Cardiac Differentiation of Human Induced Pluripotent Stem Cells Reveals Faster Cardiomyocyte Maturation Compared to 2D Culture. Sci. Rep. 2019, 9, 9229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; McKeithan, W.L.; Serrano, R.; Kitani, T.; Burridge, P.W.; del Álamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell–derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.A.; Dias, T.P.; Cabral, J.M.S.; Pinto-do-Ó, P.; Diogo, M.M. Human multilineage pro-epicardium/foregut organoids support the development of an epicardium/myocardium organoid. Nat. Commun. 2022, 13, 6981. [Google Scholar] [CrossRef] [PubMed]
- Drakhlis, L.; Biswanath, S.; Farr, C.M.; Lupanow, V.; Teske, J.; Ritzenhoff, K.; Franke, A.; Manstein, F.; Bolesani, E.; Kempf, H.; et al. Human heart-forming organoids recapitulate early heart and foregut development. Nat. Biotechnol. 2021, 39, 737–746. [Google Scholar] [CrossRef]
- Olmsted, Z.T.; Paluh, J.L. A Combined Human Gastruloid Model of Cardiogenesis and Neurogenesis. iScience 2022, 25, 104486. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| MULTILINEAGE DIFFERENTIATION | |||
|---|---|---|---|
| Reference | Drug(s)/Pollutants | Differentiation and Drug Exposure Strategy | Redouts |
| Embryoid Bodies | |||
| [37] | Thalidomide | Culture Platform: V-shaped plate (D0–D4) + Bacteriological plate suspension culture (D4–D12) Duration of Differentiation: 14 days Drug Exposure: every other day D0 to D14 Differentiation Protocol: DMEM-F12 + 20% KO serum replacement + 1% non-essential amino acids + 0.1 mM β-mercaptoethanol | Transcriptomic profile: Microarray (D14) |
| [35] | Valproic acid | Culture Platform: V-shaped plate (D0–D4) + ULA plate (D4–D14) Duration of Differentiation: 14 days Drug Exposure: every other day from D0 to D14 Differentiation Protocol: DMEM-F12 + 20% KO serum replacement + 1% non-essential amino acids | Transcriptomic profile: RNA-sequencing (D14) |
| [34] | caffeine penicillin-G valproic acid | Culture platform: AggreWell plate (D0–D3) + 24-ULA plate (D13–D12) * Duration of Differentiation: 12 days Drug Exposure: every three days from D0 to D12 Differentiation Protocol: AggreWell™ EB Formation Medium * Use of MACS purified TRA-1-60 positive hPSCs | - EBs size and shape (D3, D6, D9, D12): BF images and circularity coefficient calculation - Mesoderm and ectoderm genes expression (D12): RT-PCR for the mesodermal genes KDR, C-ACTIN and BRACHYURY; and for the ectodermal genes NETO2, NCAM, NES, BIII-TUB and NEFH |
| [39] | Nicotine | Culture Platform: Aggrewell 800 plate (D0–D1) + ULA-6well plate (D1–D21) Duration of Differentiation: 21 days Drug Exposure: every day from D0 to D21 Differentiation Protocol: DMEM/F12 + 20% FBS + 2 mM L-glutamine + 1x non-essential amino acids + 100 mM β-mercaptoethanol | - Transcriptomic profile: Single-cell RNA sequencing (D21) - EBs size: BF images - ATP Activity: CellTiter-Glo 2.0 - ROS production: ROS-Glo H2O2 |
| [38] | Valproic Acid All-trans Retinoic Acid Thalidomide Methotrexate Hydroxyurea Ascorbic acid Penicillin G Ibuprofen | Culture Platform: 96-Well Plate Duration of Differentiation: 7 days Drug Exposure: drug treatment on D0, D3 and D5 Differentiation Protocol: DMEM + 20% KO-SR + 1% GlutaMAX + 1% non-essential aminoacids + 0.18% β-mercaptoethanol, | Expression of 96 pre-selected developmentally genes: RT-PCR |
| [36] | folic acid all-trans retinoic acid dexamethasone valproic acid | Culture Platform: ULA plate (D0–D5) + 2D Matrigel (D5–D15) Duration of Differentiation: 15 days Drug exposure: every two days from D0 to D15 Differentiation Protocol: mTeSR1 | Transcriptomic profile: RNA-sequencing (D15) |
| [40] | Cadmium | Culture Platform: 384-well plate Culture Duration: 8 days Drug exposure: every other day from D1 to D8 Differentiation Protocol: E6 medium | - Percentage of cTnT positive cells (D8): Flow Cytometry - Gene expression (D8) |
| Gastruloids | |||
| [41] | All-trans retinoic acid Valproic Acid Bosentan Thalidomide Phenytoin Ibuprofen Penicillin G | Culture format: 96-Well plate Culture Duration: 72 h Drug exposure: 0–24 h Differentiation Protocol: 0–24 h: CHIR | - Morphological shape descriptors (circularity, lack of elongation, size): BF images - Fluorescence analysis: use of ES report cell line RUES2- GLR (mCit–SOX2—neuroectoderm, mCerulean–BRA—mesoderm, tdTomato–SOX17—endoderm) |
| [42] | Remdesivir | Culture format: 96-well plate Culture Duration: 5 days Drug exposure: single addition at D0 without medium change Differentiation Protocol: CHIR + SB431542 + retinoic acid | - Gastruloid morphologic analyses (area, elongation distortion index, and aspect ratio) (D5): BF images - Gene expression (D5): RT-PCR for the somites genes MEOX1, MESP2, PAX3); the cranial caudal axis genes ALDH1A2, FGF8, HOXB7, HOXB9, WNT5A; and for the neuroectoderm genes NEUROG2, OLIG3, PAX6 |
| MESENDODERM/CARDIAC DIFFERENTIATION | |||
|---|---|---|---|
| Reference | Drug(s)/Pollutants | Differentiation and Drug Exposure Strategy | Redouts |
| 2D Culture | |||
| [43] | 71 drug-like compounds | Differentiation Type: Mesendoderm commitment Culture Platform: 96-well Plate Duration of Differentiation: 3 days Drug Exposure: D0 and D1 Differentiation Protocol: D0–D1: RPMI + 2 mM L-glutamine + Activin A + WNT3A D1–D3: RPMI + L-glutamine + Activin A + 0.1% FBS | SOX17 protein expression (D3): Antibody staining (Counting the number of SOX17+ nuclei within the total DAPI+ nuclei) |
| [44,45] | 30 drug-like compounds | Differentiation Type: Mesoderm commitment Culture Platform: micropatterned circular molds Duration of Differentiation: 3 days Drug Exposure: Single administration at D0(?) Differentiation Protocol: D0–D3: Activin A, BMP4, FGF2 | Brachyury (T) protein expression (D3): Antibody staining (4 analyzed parameters: area of the T+ region; relative distance of the T+ region to the colony centroid and outline; standard deviation of the distribution of the T+ region; coefficient of variation of the distribution of the T+ region) |
| [46] | 13-cis-retinoic acid (Isotretinoin) | Differentiation Type: CM differentiation Culture Platform: 12-well plate Duration of Differentiation: 6 days Drug Exposure: D0–D6 Differentiation Protocol: Gibco™ PSC Cardiomyocyte Differentiation Kit | Transcriptomic profile: RNA sequencing and ATAC sequencing (D0, D2 and D6): |
| [47] | Arsenic Trioxide | Differentiation Type: CM differentiation Culture Platform: Matrigel-coated plates Duration of Differentiation: >4 days Drug Exposure: compound was added at D0–D1 or D0–D2 Differentiation Protocol: D0–D2: differentiation basal medium I (CELLAPY) D2–D4: differentiation basal medium II (CELLAPY) D4-: differentiation basal medium III (CELLAPY) | - Cell Death: TUNEL assay (24 h of drug exposure) (D2, D4, D6) - Cell Proliferation: EdU staining (24 h of drug exposure) (D2, D4, D6) - Gene expression: RT-PCR for EOMES and T (24 h and 48 h of drug exposure) (D2); GATA4, MESP1, TBX5 (24 h of drug exposure) (D4); and α-ACTININ (24 h and 48 h of drug exposure) (D6?) - DNA damage: Immunostaining for γH2AX (24 h of drug exposure) (D2) |
| [48] | Dioxin | Differentiation Type: CM differentiation Culture Platform: Matrigel-coated plates Duration of Differentiation: 14 days Drug Exposure: From D-3 to D12 of differentiation (every time that the medium was changed) (Tested different exposure setups D-3–D0; D1–D4; D5–D8; D9–D14; D–3–D14) Differentiation Protocol: D0–D2: CHIR D2–D4: IWR-1 | First assessment for all 5 tested conditions: - Gene expression (D14): RT-PCR for TNNT2, ACTN2 and MYL2 - Immunostaining for ACTN2 (D14) Drug exposure from (D-3–D0) - Gene expression: RT-PCR for T and GSC (D2); ISL1 and TBX5 (D5); TNNT2 and NKX2.5 (D8); ACTN2 and TNNT2 (D14) 2. Immunostaining for T (D2), ISL1 (D5), TNNT2 and ACTN2 (D8, D14) 3. Transcriptomic profile (D2): RNA-sequencing |
| [49] | Ribavirin | Differentiation Type: CM differentiation Culture Platform: Matrigel-coated plates Duration of Differentiation: 7 days Drug Exposure: D0, D1, D3, D5 (Tested different drug exposure setups: From 1. hPSC to mesoderm; 2. mesoderm to cardiac progenitors; 3. cardiac progenitors to cardiomyocytes) Differentiation Protocol: D0–D1: CHIR D3–D5: IWR-1 | - Contraction (visual beat score): Frequency of contraction and beating colonies - Cell Proliferation: EdU staining - ROS content: DCFH-DA staining - Gene expression: RT-PCR for EOMES and T (mesoderm); GATA4 and ISL1 (cardiac progenitors); cTnT and α-MHC (cardiomyocytes) |
| [50] | Drugs used for platform validation: Thalidomide Valproic acid Folic acid Saccharin Additional 17 drugs for platform testing | Differentiation Type: CM differentiation Culture Platform: Matrigel-coated 24-well plates Duration of Differentiation: 14 days Drug Exposure: D0, D3, D7, D10 Differentiation Protocol: D0–D3: CHIR + BMP4 + ActivinA D3–D5: XAV939 | - Primary redout: gene expression of BMP4 (D7) and MYH6 (D14) - Secondary redout (only used to further support the first one): Contraction (visual beat score) |
| [40] | Cadmium | Differentiation Type: CM differentiation Culture Platform: Matrigel-coated plates Duration of Differentiation: 8 days Drug Exposure: Every other day from D1 to D8 (Tested different drug exposure setups (D0–D2 or D2–D4)) Differentiation Protocol: STEMdiffTM Cardiomyocyte Differentiation Kit | - NKX2.5-positive cells (D8): Flow Cytometry (NKX2.5-GFP reporter cell line) - Gene expression: RT-PCR for MESP1, EOMES, MIXL1, HAND1, SNAI2 and HOPX (D2); and NKX2–5, GATA4, TNNT2, α-actinin (D8) - Histone methylation (H3K27 trimethylation (H3K27me3) and H3K4 trimethylation (H3K4me3)) (D2): Western Blot |
| [51] | 5-Fluorouracil Penicillin G | Differentiation Type: CM differentiation Culture Platform: Matrigel-coated 24-well plates Duration of Differentiation: 10 days Drug Exposure: D2, D3, D4, D6, and D8 Differentiation Protocol: D0–D1: CHIR + BMP4 + FGF2 D2–D4: IWP2 | - Contraction (D10): BF video (frequency of contraction and contraction area) - Gene expression (D10): RT-PCR for TNNT2 and ACTN2 |
| 3D Culture | |||
| [31] | Thalidomide Valproic acid Epoxiconazole | Model: CM aggregates Culture Platform: ULA 96-well plate Duration of Differentiation: 7 Days Drug Exposure: D1, D2, D3, D6 (do not expose at D0) Differentiation Protocol: D0–D1: CHIR + BMP4 + ActivinA + FGF2 D2–D3: WNT-C59 | - Contraction (D7): Visual beat score (if no movement was seen, the beat score 0 was given; if the entire area of the sphere contracted, a beat score of 2 was given; everything in between was given a score of 1) - Aggregate size (D7): BF images |
| [52] | Thalidomide Valproic acid | Model: CM aggregates Culture Platform: ULA 96-well plate Duration of Differentiation: 7 Days Drug Exposure: D1, D2, D3, D6 (do not expose at D0) Differentiation Protocol: D0–D1: CHIR + BMP4 + ActivinA + FGF2 D2–D3: WNT-C59 | - Contraction (D7): BF video - NKX2.5 expression level (D7): Luminescence measurement using a NKX2.5-reporter line |
| [54] | Doxylamine Succinate Amoxicillin Rifampicin Lithium Carbonate Phenytoin Doxycycline All-trans-RA (Tretinoin) 13-cis-RA (Isotretinoin) Thalidomide | Model: Cardiac Organoid Culture Platform: Micropatterned platform Duration of Differentiation: 20 Days Drug Exposure: D1, D2, D4, D6–D20 (do not expose at D0) Differentiation Protocol: D0–D1: CHIR D2–D4: IWP4 | - Percentage of cTnT-positive cells (D20): Flow cytometry - cTnT-positive area (D20): Area ratio - 3D tissue morphology (D20) - Contraction (D20): BF video and calcium transient profile (hPSC line engineered with a calcium sensor GCaMP6f) (Contraction velocity and BPM) |
| [53] | Aspirin Thalidomide Retinoid Plastic residuals | Model: AVC; LV and RV. OFT, atrial organoids Culture Platform: 96-well plate Duration of differentiation: 9.5 days | - Gene expression (D3.5, D4.5 and D9.5) - Morphology (D3.5, D4.5 and D9.5): BF image - Patterning (D3.5, D4.5 and D9.5): TNNI1-GFP reporter cell line |
| [40] | Cadmium | Model: Cardiac Organoids (atrial and ventricle CMs, CFs, endocardial and endothelial cells) Culture Platform: Culture Duration: 8 days Drug Exposure: every other day from D1 to D8 Differentiation Protocol: D0–D1: CHIR + BMP4 + Activin A D2–D4: IWR-1 | - Contraction (D8) - Gene expression (D8): RT-PCR for selected genes associated with cardiomyocytes, endocardial cells, and cardiac fibroblasts |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, M.A.; Nunes, T.C.; Cabral, J.M.S.; Diogo, M.M. Developmental Toxicity Studies: The Path towards Humanized 3D Stem Cell-Based Models. Int. J. Mol. Sci. 2023, 24, 4857. https://doi.org/10.3390/ijms24054857
Branco MA, Nunes TC, Cabral JMS, Diogo MM. Developmental Toxicity Studies: The Path towards Humanized 3D Stem Cell-Based Models. International Journal of Molecular Sciences. 2023; 24(5):4857. https://doi.org/10.3390/ijms24054857
Chicago/Turabian StyleBranco, Mariana A., Tiago C. Nunes, Joaquim M. S. Cabral, and Maria Margarida Diogo. 2023. "Developmental Toxicity Studies: The Path towards Humanized 3D Stem Cell-Based Models" International Journal of Molecular Sciences 24, no. 5: 4857. https://doi.org/10.3390/ijms24054857