
NFIXing Cancer: The Role of NFIX in Oxidative Stress Response and Cell Fate
 , ,
, ,
Abstract
:1. Introduction
2. NFIX Roles in Development
3. Roles of NFIX in Cancer
3.1. NFIX and Oxidative Stress
3.2. NFIX and Cell Fate
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| ADAM10 | Disintegrin and metalloproteinase domain-containing protein 10 |
| ATF3 | Activating transcription factor 3 |
| BCL2 | B-cell leukemia/lymphoma 2 |
| Bcl2l1/BCL-XL | Bcl-2-like protein 1 or B-cell lymphoma-extra large |
| c-MLP | Thrombopoietin receptor or myeloproliferative leukemia protein |
| ceRNAs | Competing endogenous RNAs |
| circRNA | Circular RNA |
| CKM | Muscle creatine kinase |
| CYP1A1 | Cytochrome P450 1A1 |
| EMX2 | Empty spiracles homolog 2 |
| E2A | Immunoglobulin enhancer-binding factors |
| EBF | Early B cell factor |
| ENO3 | β-enolase |
| ERK | Extracellular signal-regulated kinases |
| EZR | Ezrin |
| GDNF | Glial cell derived neurotrophic factor |
| IL6ST | Interleukin-6 receptor subunit β |
| HES | Hairy and enhancer of split-1 |
| HOX | Homeobox genes |
| Insc | Inscuteable |
| ITGB1 | Integrin β-1 |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription |
| JUNB | AP-1 transcription factor subunit |
| lncRNA | Long non-coding RNA |
| MHC | Myosin heavy chain |
| MAL | Megakaryocytic acute leukemia |
| MAPK | Mitogen-activated protein kinase |
| MASH1 | Homologue of ASCL1, Achaete-scute homolog 1 |
| MAST1 | Microtubule Associated Serine/Threonine Kinase 1 |
| miRNA | microRNA |
| NFI | Nuclear factor I |
| NFIX | Nuclear factor I X |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NURF | Nucleosome remodeling factor |
| PAX6/7 | Paired box gene 6/7 |
| PI3K/AKT | Phosphoinositide 3-kinase/protein kinase B |
| Pir | Pirin |
| PKN1 | Protein Kinase N1 |
| RAC1/2 | Rac Family Small GTPase 1/2 |
| RhoA | Ras Homolog Family Member A |
| RPN2 | Ribophorin-II |
| ROCK | Rho associated coiled-coil containing protein kinase 1 |
| ROS | Reactive oxygen species |
| SRF | Serum response factor |
| STAT3/6 | Signal transducer and activator of transcription 3/6 |
| SOX | SRY-Box Transcription Factor |
| TGF-β | Transforming growth factor-beta |
| TIMP1 | Metalloproteinase inhibitor 1 |
References
- Gronostajski, R.M. Roles of the NFI/CTF gene family in transcription and development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Piper, M.; Gronostajski, R.; Messina, G. Nuclear factor one X in development and disease. Trends Cell Biol. 2018, 29, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Kruse, U.; Lichter, P.; Sippel, A.E. Chromosomal localization of the four genes (NFIA, B, C, and X) for the human transcription factor nuclear factor I by fish. Genomics 1995, 28, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kruse, U.; Sippel, A.E. Transcription factor nuclear factor I proteins form stable homo- and heterodimers. FEBS Lett. 1994, 348, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göös, H.; Kinnunen, M.; Salokas, K.; Tan, Z.; Liu, X.; Yadav, L.; Zhang, Q.; Wei, G.-H.; Varjosalo, M. Human transcription factor protein interaction networks. Nat. Commun. 2022, 13, 1–16. [Google Scholar] [CrossRef]
- Fraser, J.; Essebier, A.; Brown, A.S.; Davila, R.A.; Harkins, D.; Zalucki, O.; Shapiro, L.P.; Penzes, P.; Wainwright, B.J.; Scott, M.P.; et al. Common regulatory targets of NFIA, NFIX and NFIB during postnatal cerebellar development. Cerebellum 2019, 19, 89–101. [Google Scholar] [CrossRef]
- Chen, K.-S.; Lim, J.W.; Richards, L.J.; Bunt, J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett. 2017, 410, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Steele-Perkins, G.; Butz, K.G.; Lyons, G.E.; Zeichner-David, M.; Kim, H.-J.; Cho, M.-I.; Gronostajski, R.M. Essential role for NFI-C/CTF transcription-replication factor in tooth root development. Mol. Cell. Biol. 2003, 23, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Messina, G.; Biressi, S.; Monteverde, S.; Magli, A.; Cassano, M.; Perani, L.; Roncaglia, E.; Tagliafico, E.; Starnes, L.; Campbell, C.E.; et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell 2010, 140, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.; Genovesi, L.A.; Gronostajski, R.M.; Wainwright, B.J.; Piper, M. Nuclear factor one transcription factors: Divergent functions in developmental versus adult stem cell populations. Dev. Dyn. 2014, 244, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Fane, M.; Harris, L.; Smith, A.G.; Piper, M. Nuclear factor one transcription factors as epigenetic regulators in cancer. Int. J. Cancer 2017, 140, 2634–2641. [Google Scholar] [CrossRef] [Green Version]
- Alevizopoulos, A.; Dusserre, Y.; Tsai-Pflugfelder, M.; von der Weid, T.; Wahli, W.; Mermod, N. A proline-rich TGF-beta-responsive transcriptional activator interacts with histone H3. Genes Dev. 1995, 9, 3051–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusserre, Y.; Mermod, N. Purified cofactors and histone H1 mediate transcriptional regulation by CTF/NF-I. Mol. Cell. Biol. 1992, 12, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- Pjanic, M.; Schmid, C.D.; Gaussin, A.; Ambrosini, G.; Adamcik, J.; Pjanic, P.; Plasari, G.; Kerschgens, J.; Dietler, G.; Bucher, P.; et al. Nuclear Factor I genomic binding associates with chromatin boundaries. BMC Genom. 2013, 14, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denny, S.K.; Yang, D.; Chuang, C.-H.; Brady, J.J.; Lim, J.S.; Grüner, B.M.; Chiou, S.-H.; Schep, A.N.; Baral, J.; Hamard, C.; et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 2016, 166, 328–342. [Google Scholar] [CrossRef] [Green Version]
- Pjanic, M.; Pjanic, P.; Schmid, C.; Ambrosini, G.; Gaussin, A.; Plasari, G.; Mazza, C.; Bucher, P.; Mermod, N. Nuclear factor I revealed as family of promoter binding transcription activators. BMC Genom. 2011, 12, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker-Santos, D.D.; Lonergan, K.M.; Gronostajski, R.M.; Lam, W.L. Nuclear factor I/B: A Master regulator of cell differentiation with paradoxical roles in cancer. EBioMedicine 2017, 22, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.; Piper, M.; Gronostajski, R.M.; Richards, L.J. Nuclear factor one transcription factors in cns development. Mol. Neurobiol. 2008, 39, 10–23. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Gronostajski, R.M. Identification of a conserved oxidation-sensitive cysteine residue in the NFI family of DNA-binding proteins. J. Biol. Chem. 1994, 269, 29949–29955. [Google Scholar] [CrossRef]
- Liu, S.; Qu, D.; Li, W.; He, C.; Li, S.; Wu, G.; Zhao, Q.; Shen, L.; Zhang, J.; Zheng, J. miR-647 and miR-1914 promote cancer progression equivalently by downregulating nuclear factor IX in colorectal cancer. Mol. Med. Rep. 2017, 16, 8189–8199. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yang, Z.; Yao, R.; Li, Y.; Liu, Z.; Chen, X.; Zhang, G. miR-210 promotes progression of endometrial carcinoma by regulating the expression of NFIX. Int. J. Clin. Exp. Pathol. 2018, 11, 5213–5222. [Google Scholar]
- Kleemann, M.; Schneider, H.; Unger, K.; Sander, P.; Schneider, E.M.; Fischer-Posovszky, P.; Handrick, R.; Otte, K. MiR-744-5p inducing cell death by directly targeting HNRNPC and NFIX in ovarian cancer cells. Sci. Rep. 2018, 8, 9020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yuan, Q.; Zhang, W.; Niu, M.; Fu, H.; Qiu, Q.; Mao, G.; Wang, H.; Wen, L.; Wang, H.; et al. MiR-663a Stimulates proliferation and suppresses early apoptosis of human spermatogonial stem cells by targeting NFIX and regulating cell cycle. Mol. Ther. Nucleic Acids 2018, 12, 319–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of MicroRNA-223 and transcription factors NFI-A and C/EBPα regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Liu, J.; Zhang, D.; Li, B. MiR-1290 promotes cancer progression by targeting nuclear factor I/X(NFIX) in esophageal squamous cell carcinoma (ESCC). Biomed. Pharmacother. 2015, 76, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, Y.; Qi, L.; Ding, L.; Jiang, H.; Yu, H. NFIX circular RNA promotes glioma progression by regulating miR-34a-5p via notch signaling pathway. Front. Mol. Neurosci. 2018, 11, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veno, M.T.; Hansen, T.B.; Venø, S.T.; Clausen, B.H.; Grebing, M.; Finsen, B.; Holm, I.E.; Kjems, J. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015, 16, 245. [Google Scholar] [CrossRef] [Green Version]
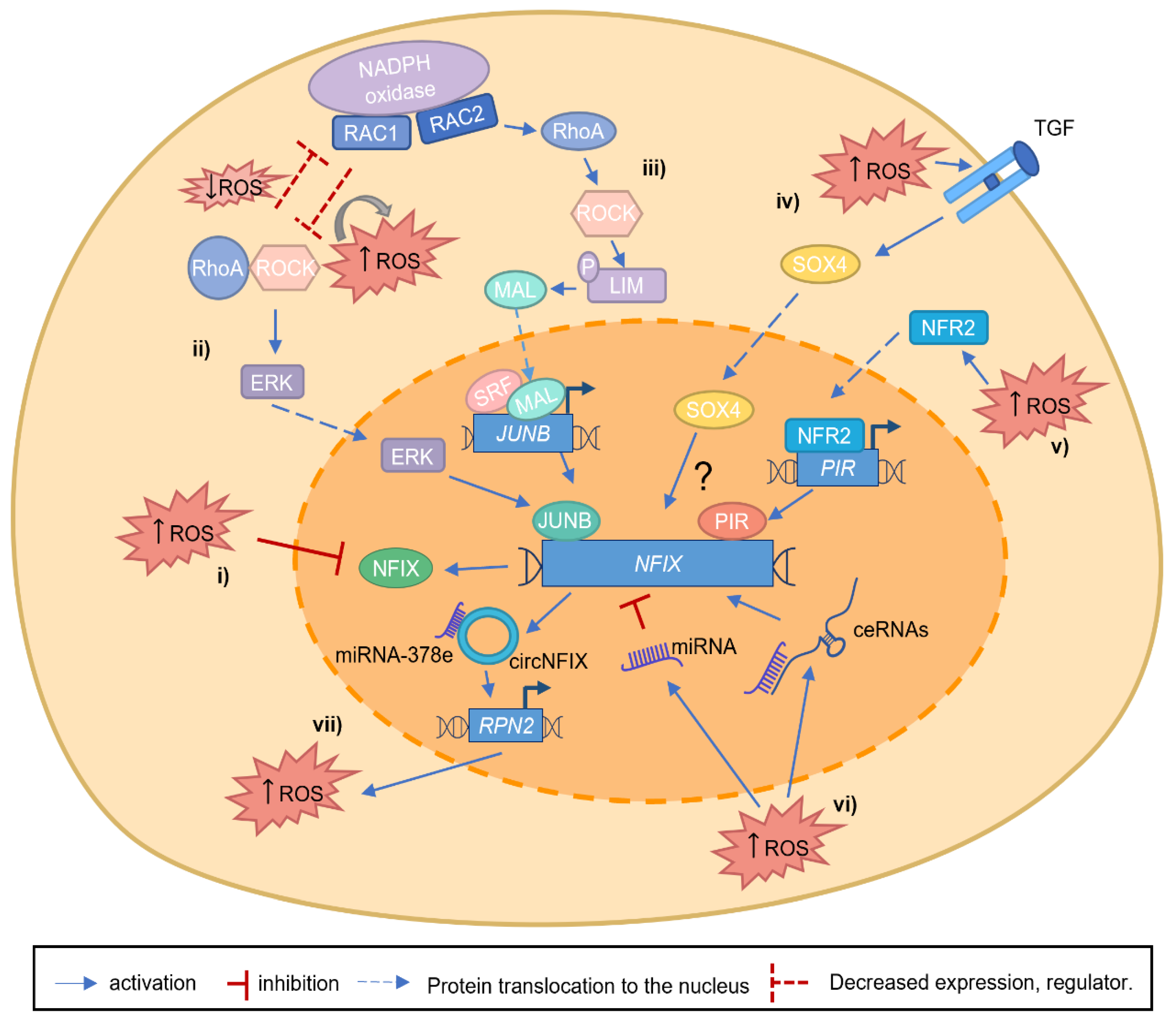
- Cui, X.; Dong, Y.; Li, M.; Wang, X.; Jiang, M.; Yang, W.; Liu, G.; Sun, S.; Xu, W. A circular RNA from NFIX facilitates oxidative stress-induced H9c2 cells apoptosis. Vitr. Cell. Dev. Biol. Anim. 2020, 56, 715–722. [Google Scholar] [CrossRef]
- Zhao, L.; Song, X.; Guo, Y.; Ding, N.; Wang, T.; Huang, L. Long non-coding RNA SNHG3 promotes the development of non-small cell lung cancer via the miR-1343-3p/NFIX pathway. Int. J. Mol. Med. 2021, 48, 1–12. [Google Scholar] [CrossRef]
- Ye, L.; Feng, W.; Weng, H.; Yuan, C.; Liu, J.; Wang, Z. MAFG-AS1 aggravates the progression of pancreatic cancer by sponging miR-3196 to boost NFIX. Cancer Cell Int. 2020, 20, 591. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Starke, D.W.; Mieyal, J.J.; Gronostajski, R.M. Thioltransferase (glutaredoxin) reactivates the DNA-binding activity of oxidation-inactivated nuclear factor I. J. Biol. Chem. 1998, 273, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzo, G. Similarities between embryo development and cancer process suggest new strategies for research and therapy of tumors: A new point of view. Front. Cell Dev. Biol. 2019, 7, 20. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, P.; Wang, F.; Yang, J.; Yang, Z.; Qin, H. The relationship between early embryo development and tumourigenesis. J. Cell. Mol. Med. 2010, 14, 2697–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreesen, O.; Brivanlou, A.H. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Taglietti, V.; Angelini, G.; Mura, G.; Bonfanti, C.; Caruso, E.; Monteverde, S.; Le Carrou, G.; Tajbakhsh, S.; Relaix, F.; Messina, G. Rhoa and erk signalling regulate the expression of the transcription factor nfix in myogenic cells. Development 2018, 145, dev163956. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Taglietti, V.; Messina, G. Targeting Nfix to fix muscular dystrophies. Cell Stress 2018, 2, 17–19. [Google Scholar] [CrossRef] [Green Version]
- Rando, T.A. Oxidative stress and the pathogenesis of muscular dystrophies. Am. J. Phys. Med. Rehabil. 2002, 81, S175–S186. [Google Scholar] [CrossRef]
- Moore, T.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Martins, S.G.; Zilhão, R.; Thorsteinsdóttir, S.; Carlos, A.R. Linking Oxidative stress and DNA Damage to changes in the expression of extracellular matrix components. Front. Genet. 2021, 12, 1279. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Antonini, S.; Bonfanti, C.; Monteverde, S.; Vezzali, C.; Tajbakhsh, S.; Cossu, G.; Messina, G. Nfix Regulates temporal progression of muscle regeneration through modulation of myostatin expression. Cell Rep. 2016, 14, 2238–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saclier, M.; Lapi, M.; Bonfanti, C.; Rossi, G.; Antonini, S.; Messina, G. The transcription factor Nfix requires RhoA-ROCK1 dependent phagocytosis to mediate macrophage skewing during skeletal muscle regeneration. Cells 2020, 9, 708. [Google Scholar] [CrossRef] [Green Version]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; Van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; De Mori, R.; Baeza-Raja, B.; de Lagrán, M.M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68, 81–93. [Google Scholar] [CrossRef]
- Smith, L.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68, 602–615. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. Shifts in macrophage cytokine production drive muscle fibrosis. Nat. Med. 2015, 21, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.-K.; Lee, S.T.; Fiore, D.; Zhang, R.-H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M.V. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.Z.; Lyons, G.E.; Gronostajski, R.M. Expression patterns of the four nuclear factor I genes during mouse embryogenesis indicate a potential role in development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 1997, 208, 313–325. [Google Scholar] [CrossRef]
- Harris, L.; Zalucki, O.; Gobius, I.; McDonald, H.; Osinki, J.; Harvey, T.J.; Essebier, A.; Vidovic, D.; Gladwyn-Ng, I.; Burne, T.H.; et al. Transcriptional regulation of intermediate progenitor cell generation during hippocampal development. Development 2016, 143, 4620–4630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matuzelski, E.; Bunt, J.; Harkins, D.; Lim, J.W.; Gronostajski, R.M.; Richards, L.J.; Harris, L.; Piper, M. Transcriptional regulation of Nfix by NFIB drives astrocytic maturation within the developing spinal cord. Dev. Biol. 2017, 432, 286–297. [Google Scholar] [CrossRef]
- Heng, Y.H.E.; McLeay, R.C.; Harvey, T.J.; Smith, A.G.; Barry, G.; Cato, K.; Plachez, C.; Little, E.; Mason, S.; Dixon, C.; et al. NFIX Regulates neural progenitor cell differentiation during hippocampal morphogenesis. Cereb. Cortex 2012, 24, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.E.; Wynn, S.L.; Sesay, A.; Cruz, C.; Cheung, M.; Gaviro, M.V.G.; Booth, S.; Gao, B.; Cheah, K.S.; Lovell-Badge, R.; et al. SOX9 induces and maintains neural stem cells. Nat. Neurosci. 2010, 13, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Heng, Y.H.E.; Zhou, B.; Harris, L.; Harvey, T.; Smith, A.; Horne, E.; Martynoga, B.; Andersen, J.; Achimastou, A.; Cato, K.; et al. NFIX Regulates proliferation and migration within the murine SVZ neurogenic niche. Cereb. Cortex 2014, 25, 3758–3778. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Campos, J.; Osinski, J.M.; Gronostajski, R.M.; Michie, A.M.; Keeshan, K. Nfix Expression critically modulates early B lymphopoiesis and myelopoiesis. PLoS ONE 2015, 10, e0120102. [Google Scholar] [CrossRef]
- Singh, H.; Medina, K.L.; Pongubala, J.M.R. Contingent gene regulatory networks and B cell fate specification. Proc. Natl. Acad. Sci. USA 2005, 102, 4949–4953. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.; Walker, M.; Ganuza, M.; Holmfeldt, P.; Bordas, M.; Kang, G.; Bi, W.; Palmer, L.E.; Finkelstein, D.; McKinney-Freeman, S. Nfix promotes survival of immature hematopoietic cells via regulation of C-MPL. Stem Cells 2018, 36, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Davila, R.A.; Spiller, C.; Harkins, D.; Harvey, T.; Jordan, P.W.; Gronostajski, R.M.; Piper, M.; Bowles, J. Deletion of NFIX results in defective progression through meiosis within the mouse testis. Biol. Reprod. 2022, 106, 1191–1205. [Google Scholar] [CrossRef]
- Landi, S.; Barbuti, A.F. The Transcription Factor Nfix as a Novel Modulator of Heart Rate. Ph.D. Thesis, The University of Milan, Milano, Italy, 2018. [Google Scholar]
- Jaé, N.; Heumüller, A.W.; Fouani, Y.; Dimmeler, S. Long non-coding RNAs in vascular biology and disease. Vasc. Pharmacol. 2019, 114, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Mester-Tonczar, J.; Hašimbegović, E.; Spannbauer, A.; Traxler, D.; Kastner, N.; Zlabinger, K.; Einzinger, P.; Pavo, N.; Goliasch, G.; Gyöngyösi, M. Circular RNAs in cardiac regeneration: Cardiac cell proliferation, differentiation, survival, and reprogramming. Front. Physiol. 2020, 11, 580465. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Xu, Z.; Guo, G.; Xu, C.; Song, Z.; Li, K.; Zhong, K.; Wang, D. Circ_nuclear factor I X (circNfix) attenuates pressure overload-induced cardiac hypertrophy via regulating miR-145-5p/ATF3 axis. Bioengineered 2021, 12, 5373–5385. [Google Scholar] [CrossRef]
- Huang, S.; Li, X.; Zheng, H.; Si, X.; Li, B.; Wei, G.; Li, C.; Chen, Y.; Chen, Y.; Liao, W.; et al. Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation 2019, 139, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.W.; Lim, B.T.; Anene-Nzelu, C.G.; Ackers-Johnson, M.; Dashi, A.; See, K.; Tiang, Z.; Lee, D.P.; Chua, W.W.; Luu, T.D.; et al. A landscape of circular RNA expression in the human heart. Cardiovasc. Res. 2016, 113, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Koren, L.; Elhanani, O.; Kehat, I.; Hai, T.; Aronheim, A. Adult cardiac expression of the activating transcription factor 3, ATF3, promotes ventricular hypertrophy. PLoS ONE 2013, 8, e68396. [Google Scholar] [CrossRef] [PubMed]
- Soraya, A.-S.; Tali, H.; Rona, S.; Tom, F.; Roy, K.; Ami, A. ATF3 expression in cardiomyocytes and myofibroblasts following transverse aortic constriction displays distinct phenotypes. IJC Hear. Vasc. 2021, 32, 100706. [Google Scholar] [CrossRef]
- Victorino, P.; Marra, C.; Iacobas, D.; Iacobas, S.; Spray, D.; Linden, R.; Adesse, D.; Petrs-Silva, H. Retinal Genomic fabric remodeling after optic nerve injury. Genes 2021, 12, 403. [Google Scholar] [CrossRef]
- Yuan, B.; Cui, J.; Wang, W.; Deng, K. Gα12/13 signaling promotes cervical cancer invasion through the RhoA/ROCK-JNK signaling axis. Biochem. Biophys. Res. Commun. 2016, 473, 1240–1246. [Google Scholar] [CrossRef]
- Pei, H.; Guo, Z.; Wang, Z.; Dai, Y.; Zheng, L.; Zhu, L.; Zhang, J.; Hu, W.; Nie, J.; Mao, W.; et al. RAC2 promotes abnormal proliferation of quiescent cells by enhanced JUNB expression via the MAL-SRF pathway. Cell Cycle 2018, 17, 1115–1123. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.S. SOX4: The unappreciated oncogene. Semin. Cancer Biol. 2020, 67, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; McCabe, C.D.; Ali-Seyed, M.; Berger, M.F.; Bulyk, M.L.; Moreno, C.S. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res. 2009, 69, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, A.F.; Orlando, U.D.; López, P.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Gene expression profile and signaling pathways in MCF-7 breast cancer cells mediated by acyl-coa synthetase 4 overexpression. Transcr. Open Access 2015, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Moura, D.; Díaz-Martín, J.; Bagué, S.; Orellana-Fernandez, R.; Sebio, A.; Mondaza-Hernandez, J.; Salguero-Aranda, C.; Rojo, F.; Hindi, N.; Fletcher, C.; et al. A novel NFIX-STAT6 gene fusion in solitary fibrous tumor: A case report. Int. J. Mol. Sci. 2021, 22, 7514. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chinnaiyan, A.M. Oncogenic gene fusions in epithelial carcinomas. Curr. Opin. Genet. Dev. 2009, 19, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [Green Version]
- Kastnerova, L.; Luzar, B.; Goto, K.; Grishakov, V.; Gatalica, Z.; Kamarachev, J.; Martinek, P.; Hájková, V.; Grossmann, P.; Imai, H.; et al. Secretory carcinoma of the skin. Am. J. Surg. Pathol. 2019, 43, 1092–1098. [Google Scholar] [CrossRef]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.-L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12, R6. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.I.; Abdul Murad, N.A.; Mollah, M.M.; Jamal, R.; Harun, R. NFIX as a master regulator for lung cancer progression. Front. Pharmacol. 2017, 8, 540. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ge, R.; Zhou, J.; Yang, X.; Cheng, K.K.-Y.; Tao, J.; Wu, D.; Mao, J. Nuclear factor IX promotes glioblastoma development through transcriptional activation of Ezrin. Oncogenesis 2020, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, M.M.; Elliott, A.D.; DeGraff, D.J.; Anderson, P.D.; Anumanthan, G.; Yamashita, H.; Sun, Q.; Friedman, D.B.; Hachey, D.L.; Yu, X.; et al. NFI transcription factors interact with FOXA1 to regulate prostate-specific gene expression. Mol. Endocrinol. 2014, 28, 949–964. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.D.; Whitaker, A.M.; Schaich, M.A.; Smith, M.S.; Flynn, T.S. Base excision repair of oxidative DNA damage from mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Morel, Y.; Barouki, R. Down-regulation of cytochrome P450 1A1 gene promoter by oxidative stress. Critical contribution of nuclear factor 1. J. Biol. Chem. 1998, 273, 26969–26976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- Morel, Y.; Mermod, N.; Barouki, R. An autoregulatory loop controlling CYP1A1 gene expression: Role of H2O2 and NFI. Mol. Cell. Biol. 1999, 19, 6825–6832. [Google Scholar] [CrossRef] [Green Version]
- Taverne, Y.J.; Merkus, D.; Bogers, A.J.; Halliwell, B.; Duncker, D.J.; Lyons, T.W. Reactive oxygen species: Radical factors in the evolution of animal life. Bioessays 2018, 40, 1700158. [Google Scholar] [CrossRef]
- Rodriguez, M.; Potter, D.A. CYP1A1 regulates breast cancer proliferation and survival. Mol. Cancer Res. 2013, 11, 780–792. [Google Scholar] [CrossRef] [Green Version]
- Androutsopoulos, V.P.; Spyrou, I.; Ploumidis, A.; Papalampros, A.E.; Kyriakakis, M.; Delakas, D.; Spandidos, D.A.; Tsatsakis, A.M. Expression profile of CYP1A1 and CYP1B1 enzymes in colon and bladder tumors. PLoS ONE 2013, 8, e82487. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Liu, X.-L.; Liu, H.-S.; Luo, X.-Y.; Yuan, Y.; Ji, Y.-M.; Liu, T.; Guo, J.-L.; Zhang, J. The risk model based on the three oxidative stress-related genes evaluates the prognosis of LAC patients. Oxidative Med. Cell. Longev. 2022, 2022, 4022896. [Google Scholar] [CrossRef]
- Sun, C.; Guo, E.; Zhou, B.; Shan, W.; Huang, J.; Weng, D.; Wu, P.; Wang, C.; Wang, S.; Zhang, W.; et al. A reactive oxygen species scoring system predicts cisplatin sensitivity and prognosis in ovarian cancer patients. BMC Cancer 2019, 19, 1061. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jee, B.A.; Kwon, S.M.; Yoon, Y.; Xu, W.G.; Wang, H.; Wang, X.W.; Thorgeirsson, S.S.; Lee, J.; Woo, H.G.; et al. Identification of a mitochondrial defect gene signature reveals NUPR1 as a key regulator of liver cancer progression. Hepatology 2015, 62, 1174–1189. [Google Scholar] [CrossRef]
- Patop, I.L.; Kadener, S. circRNAs in cancer. Curr. Opin. Genet. Dev. 2018, 48, 121–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Wan, Y.; Zhao, Y.; Wen, Q.; Tang, X.; Shen, J.; Wu, X.; Li, M.; Li, X.; et al. Circular RNAs in the regulation of oxidative stress. Front. Pharmacol. 2021, 12, 1906. [Google Scholar] [CrossRef]
- Ding, C.; Wu, Z.; You, H.; Ge, H.; Zheng, S.; Lin, Y.; Wu, X.; Lin, Z.; Kang, D. CircNFIX promotes progression of glioma through regulating miR-378e/RPN2 axis. J. Exp. Clin. Cancer Res. 2019, 38, 506. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhu, Y.; Qin, Y.; Chen, Y. CircNFIX Acts as a miR-212-3p sponge to enhance the malignant progression of non-small cell lung cancer by up-regulating ADAM10. Cancer Manag. Res. 2020, 12, 9577–9587. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. A review of the molecular mechanisms of hyperglycemia-induced free radical generation leading to oxidative stress. J. Cell. Physiol. 2019, 234, 1300–1312. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Zhang, W.; Ke, Z.; Lv, Y.; Zhang, B.; Liao, Z. Ribophorin II promotes the epithelial–mesenchymal transition and aerobic glycolysis of laryngeal squamous cell carcinoma via regulating reactive oxygen species-mediated Phosphatidylinositol-3-kinase/protein kinase B activation. Bioengineered 2022, 13, 5141–5151. [Google Scholar] [CrossRef]
- Crawford, H.; Dempsey, P.; Brown, G.; Adam, L.; Moss, M. ADAM10 as a therapeutic target for cancer and inflammation. Curr. Pharm. Des. 2009, 15, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.-C.; Ni, J.-J.; Cui, W.-Y.; Wang, B.-Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar] [PubMed]
- Wang, X.; Zhou, J.; Xu, M.; Yan, Y.; Huang, L.; Kuang, Y.; Liu, Y.; Li, P.; Zheng, W.; Liu, H.; et al. A 15-lncRNA signature predicts survival and functions as a ceRNA in patients with colorectal cancer. Cancer Manag. Res. 2018, 10, 5799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmfeldt, P.; Pardieck, J.; Saulsberry, A.C.; Nandakumar, S.K.; Finkelstein, D.; Gray, J.T.; Persons, D.A.; McKinney-Freeman, S. Nfix is a novel regulator of murine hematopoietic stem and progenitor cell survival. Blood 2013, 122, 2987–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.P.; Castresana, J.S.; Shahi, M.H. Role of circular RNA in brain tumor development. Cells 2022, 11, 2130. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, C.; Liu, J.; Jiang, H.; Jiang, X.; Liu, Z. A novel tumor-promoting role for nuclear factor IX in glioblastoma is mediated through transcriptional activation of GINS1. Mol. Cancer Res. 2022, 2022, OF1–OF10. [Google Scholar] [CrossRef]
- Walker, M.; Li, Y.; Morales-Hernandez, A.; Qi, Q.; Parupalli, C.; Brown, S.A.; Christian, C.; Clements, W.K.; Cheng, Y.; Freeman, S.L.M. An NFIX-mediated regulatory network governs the balance of hematopoietic stem and progenitor cells during hematopoiesis. Blood Adv. 2022. [Google Scholar] [CrossRef]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Akbar, H.; Duan, X.; Saleem, S.; Davis, A.K.; Zheng, Y. RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive oxygen species generation in platelets. PLoS ONE 2016, 11, e0163227. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-S.; Kim, J.-G.; Jeon, C.-Y.; Won, H.-Y.; Moon, M.-Y.; Seo, J.-Y.; Kim, J.-I.; Kim, J.; Lee, J.-Y.; Choi, S.-Y.; et al. Downstream components of RhoA required for signal pathway of superoxide formation during phagocytosis of serum opsonized zymosans in macrophages. Exp. Mol. Med. 2005, 37, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazanietz, M.G.; Caloca, M.J. The rac GTPase in cancer: From old concepts to new paradigms. Cancer Res. 2017, 77, 5445–5451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Yin, C.; Huang, X.; Huang, Y.; Ding, L.; Jin, M.; Wang, Z.; Wei, J.; Li, X. ROS/TGF-β signal mediated accumulation of SOX4 in OA-FLS promotes cell senescence. Exp. Gerontol. 2021, 156, 111616. [Google Scholar] [CrossRef] [PubMed]
- Wendler, W.M.F.; Kremmer, E.; Förster, R.; Winnacker, E.-L. Identification of pirin, a novel highly conserved nuclear protein. J. Biol. Chem. 1997, 272, 8482–8489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Knatko, E.V.; Higgins, M.; Naidu, S.D.; Smith, G.; Honda, T.; de la Vega, L.; Dinkova-Kostova, A.T. Pirin, an Nrf2-regulated protein, is overexpressed in human colorectal tumors. Antioxidants 2022, 11, 262. [Google Scholar] [CrossRef]
- Brzóska, K.; Stępkowski, T.M.; Kruszewski, M. Basal PIR expression in HeLa cells is driven by NRF2 via evolutionary conserved antioxidant response element. Mol. Cell. Biochem. 2014, 389, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Perez-Dominguez, F.; Carrillo-Beltrán, D.; Blanco, R.; Muñoz, J.; León-Cruz, G.; Corvalan, A.; Urzúa, U.; Calaf, G.; Aguayo, F. Role of pirin, an oxidative stress sensor protein, in epithelial carcinogenesis. Biology 2021, 10, 116. [Google Scholar] [CrossRef]
- Miyazaki, I.; Simizu, S.; Okumura, H.; Takagi, S.; Osada, H. A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat. Chem. Biol. 2010, 6, 667–673. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shoorei, H.; Taheri, M. Non-coding RNAs are involved in the response to oxidative stress. Biomed. Pharmacother. 2020, 127, 110228. [Google Scholar] [CrossRef]
- Wang, X.; Shen, C.; Zhu, J.; Shen, G.; Li, Z.; Dong, J. Long noncoding RNAs in the regulation of oxidative stress. Oxidative Med. Cell. Longev. 2019, 2019, 1318795. [Google Scholar] [CrossRef]
- Banerjee, J.; Khanna, S.; Bhattacharya, A. MicroRNA regulation of oxidative stress. Oxidative Med. Cell. Longev. 2017, 2017, 2872156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, S.O.; Reiisi, S.; Shareef, S. miRNAs, oxidative stress, and cancer: A comprehensive and updated review. J. Cell. Physiol. 2020, 235, 8812–8825. [Google Scholar] [CrossRef] [PubMed]
- Akbari, A.; Majd, H.M.; Rahnama, R.; Heshmati, J.; Morvaridzadeh, M.; Agah, S.; Amini, S.M.; Masoodi, M. Cross-talk between oxidative stress signaling and microRNA regulatory systems in carcinogenesis: Focused on gastrointestinal cancers. Biomed. Pharmacother. 2020, 131, 110729. [Google Scholar] [CrossRef]
- Wu, L.; Du, Q.; Wu, C. CircLPAR1/miR-212-3p/ZNF217 feedback loop promotes amyloid β-induced neuronal injury in Alzheimer’s Disease. Brain Res. 2021, 1770, 147622. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Zhou, R.; Sun, J.; Zhang, F.; Tang, X.; Chen, K.K.; Zhao, J.; Lan, X.; Lin, S.; Zhang, Z.; et al. LncRNA SNHG5 promotes the progression of osteosarcoma by sponging the miR-212-3p/SGK3 axis. Cancer Cell Int. 2018, 18, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Carlos, A.R.; Weis, S.; Soares, M.P. Cross-talk between iron and glucose metabolism in the establishment of disease tolerance. Front. Immunol. 2018, 9, 2498. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omori, K.; Nakamura, A.; Miyoshi, H.; Yamauchi, Y.; Kawata, S.; Takahashi, K.; Kitao, N.; Nomoto, H.; Kameda, H.; Cho, K.Y.; et al. Glucokinase inactivation paradoxically ameliorates glucose intolerance by increasing β-cell mass in db/db Mice. Diabetes 2021, 70, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Dong, H.; Yang, Y.; Liu, B.; Zheng, M.; Cheng, Q.; Peng, L.; Li, J. NFIX downregulation independently predicts poor prognosis in lung adenocarcinoma, but not in squamous cell carcinoma. Futur. Oncol. 2018, 14, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Putative Oncogenic Gene Fusions | ||
|---|---|---|
| Type of cancer | Mechanism | References |
| Breast | NFIX-MAST1 promotes proliferation. | [77] |
| BSG-NFIX fusion present in low copy number and with unknown function. | [79] | |
| Skin | NFIX–PKN1 fusion with unknown function. | [78] |
| Sarcoma | NFIX–STAT6 fusion with unknown function. | [75] |
| Oncogene | ||
| Type of cancer | Mechanism | References |
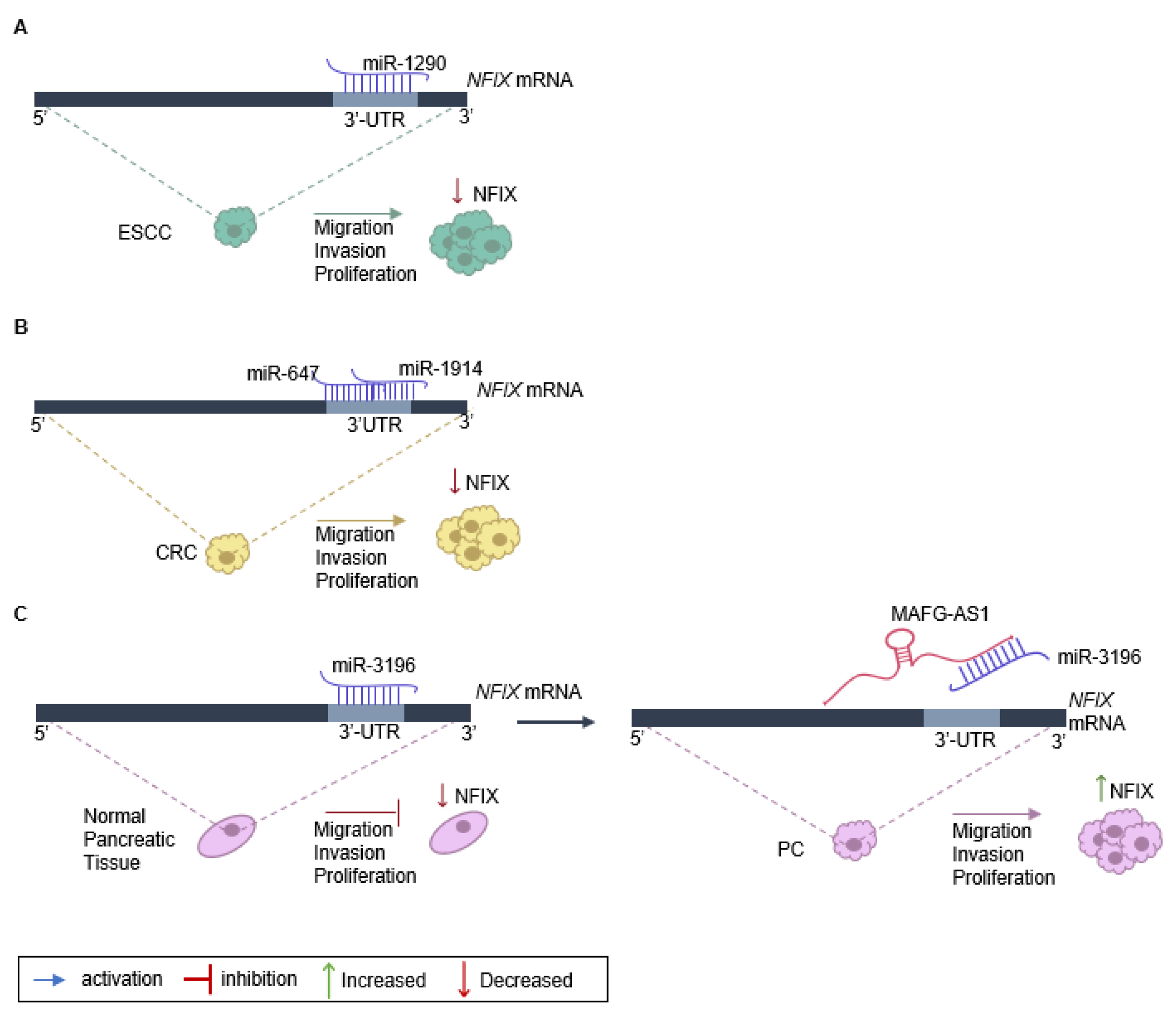
| Pancreas | ceRNA network: MAFG-AS1 binds to miR-3196 leading to NFIX expression. | [30] |
| Lung | ceRNA network: SNHG3 binds to miR-1343-3p leading to NFIX expression. | [29] |
| NFIX regulates genes involved in proliferation, migration, and invasion (IL6ST, TIMP1 and ITGB1). | [80] | |
| Brain | NFIX upregulates ezrin (EZR) promoting cell migration. | [81] |
| Prostate | NFIX binds to FOXA1 regulating prostate-specific gene expression. | [82] |
| Putative Tumor Suppressor | ||
| Type of cancer | Mechanism | References |
| Esophageal | miR-1290 binds to NFIX, decreasing its expression. | [25] |
| Colorectal | miR-647 and miR-1914 co-target NFIX, decreasing its expression. | [20] |
| Ovarian | miR-744 reduces NFIX expression, leading to apoptosis. | [22] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, V.; Martins, S.G.; Lopes, A.S.; Thorsteinsdóttir, S.; Zilhão, R.; Carlos, A.R. NFIXing Cancer: The Role of NFIX in Oxidative Stress Response and Cell Fate. Int. J. Mol. Sci. 2023, 24, 4293. https://doi.org/10.3390/ijms24054293
Ribeiro V, Martins SG, Lopes AS, Thorsteinsdóttir S, Zilhão R, Carlos AR. NFIXing Cancer: The Role of NFIX in Oxidative Stress Response and Cell Fate. International Journal of Molecular Sciences. 2023; 24(5):4293. https://doi.org/10.3390/ijms24054293
Chicago/Turabian StyleRibeiro, Vanessa, Susana G. Martins, Ana Sofia Lopes, Sólveig Thorsteinsdóttir, Rita Zilhão, and Ana Rita Carlos. 2023. "NFIXing Cancer: The Role of NFIX in Oxidative Stress Response and Cell Fate" International Journal of Molecular Sciences 24, no. 5: 4293. https://doi.org/10.3390/ijms24054293