
Whole-Genome Duplication and Genome Instability in Cancer Cells: Double the Trouble

Abstract
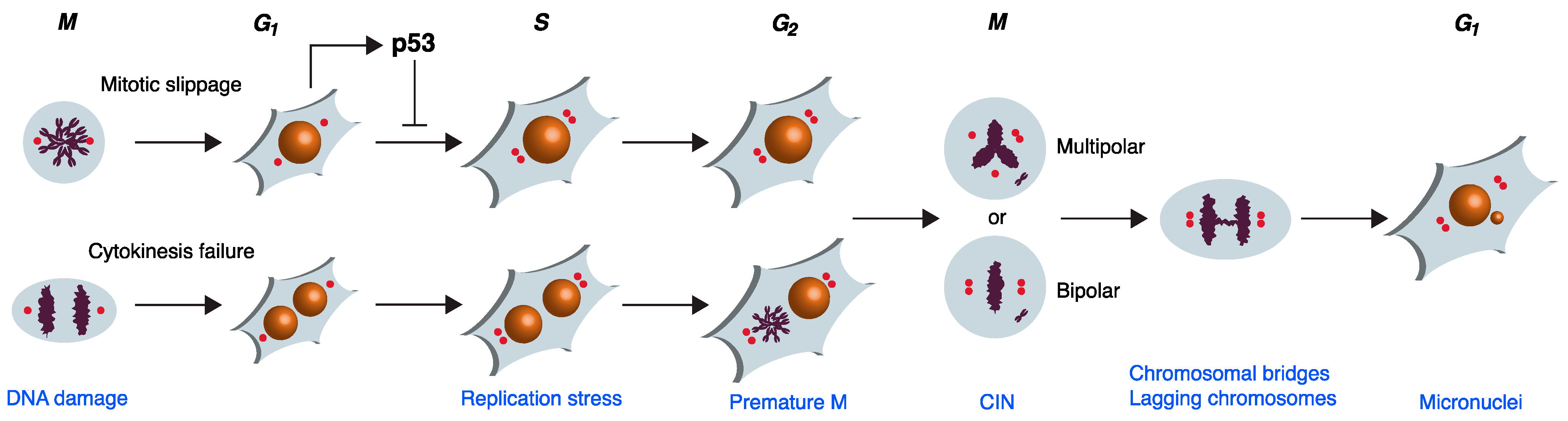
:1. Introduction: Whole-Genome Duplication in Normal and Cancer Cells
2. Mitotic Failures Leading to WGD
3. Overcoming G1 Checkpoint Arrest after Mitotic Failure
4. Genome Instability during the First Interphase after Mitotic Failure
5. Mechanisms for Mitigating Supernumerary Centrosomes after WGD

{kind=link}
| Processes | Proteins | Centrosome Clustering | RNAi Screens | References | Processes | Proteins | Centrosome Clustering | RNAi Screens | References |
|---|---|---|---|---|---|---|---|---|---|
| Actin-related | ARP3 | + | K | CENP-T | + | L | |||
| Cofilin | − | [77] | INCENP | + | G, L | ||||
| LIMK2 | + | D | MAD2 | + | K | [64] | |||
| MYHK-IIb | + | D | MPS1 | + | |||||
| MYLK | + | D | NDC80 | + | L | ||||
| MYO9B | + | D | RCC1 | + | K | ||||
| MYO10 | + | [78] | SKA1 | + | D | ||||
| MYO15 | + | K | SKA2 | + | D | ||||
| RhoA | + | K | SKA3 | + | D | ||||
| TESK1 | + | D | SPC24 | + | L | ||||
| WASL | + | K | SPC25 | + | L | ||||
| Augmin | HAUS1 | + | L | SPINDLY | + | D | |||
| HAUS3 | + | L | Survivin | + | L | ||||
| HAUS6 | + | L | [75,79] | Microtubule-related | ASPM | + | K | ||
| Centrosome | AURKA | + | [80] | CKAP5/ch-TOG | + | K, L | [81] | ||
| Calmodulin-1 | + | K, D | CLIP-170 | + | K | ||||
| Calmodulin-3 | + | K, D | FES | + | D | ||||
| CEP110 | + | [82] | HURP | + | [83] | ||||
| CEP164 | + | L | TACC | + | K | [81] | |||
| CEP215 | + | [74] | MAST1 | + | D | ||||
| CPAP | + | [67] | MAST4 | + | D | ||||
| HSP70 | + | [84] | TPX2 | + | L | ||||
| NEK6 | + | [84] | Tubulin | + | K, L | ||||
| NuMA | − | [69] | Miscellaneous | CERT1 | + | D | |||
| PLK4 | + | D | KCC2G | + | D | ||||
| Cell adhesion | CDH11 | + | L | IFT52 | + | [85] | |||
| DDR2 | + | D | IFT88 | + | [85] | ||||
| E-cadherin | − | [86] | LGN | + | [78] | ||||
| Keratin-5 | − | [87] | MLKL | + | D | ||||
| MPP2 | + | D | PRPS1 | + | D | ||||
| MPP3 | + | D | TSSK6 | + | D | ||||
| PEAK1 | + | D | Signaling | CAMK2α | + | D | |||
| Periostin | + | L | CAMK2δ | + | K | ||||
| Plakoglobin | − | [87] | CDK1/2 | + | [88] | ||||
| Tenascin-X | + | L | ILK | + | [81] | ||||
| VE-cadherin | + | L | LATS1 | + | D | ||||
| Chromatid cohesion | SCC4 | + | D | MAP3K7 | + | D | |||
| SGO1 | + | L | MAP3K11 | + | D | ||||
| Sororin | + | L | MAP4K5 | + | L | ||||
| Cytokinesis | Anillin | + | L | MARK3 | + | K, D | |||
| ECT2 | + | L | MASTL | + | D | ||||
| PRC1 | + | L | MERTK | + | D | ||||
| DNA damage response | ATM | + | D | [89] | p53 | + | [90] | ||
| ATR | + | [89] | PIK3Cβ | + | K | ||||
| NEK4 | + | D | PIP4K2β | + | D | ||||
| PARP1 | + | PIP4K2γ | + | D | |||||
| PARP6 | + | [91] | PP2A-Aα | − | [92] | ||||
| Dynein-dynactin | ARP1 | + | D | STAT3 | + | [93] | |||
| DYNC1H1 | + | D, L | STK10 | + | D | ||||
| DYNC1I2 | + | D | STK33 | + | D | ||||
| Dynein | + | [69] | TAOK1 | + | D | ||||
| DYNLRB1 | + | D | TAOK2 | + | D | ||||
| LIS1 | + | D | TRPM7 | + | D | ||||
| p22 | + | D | TSSK2 | + | D | ||||
| p150Glued | + | D | TSSK3 | + | D | ||||
| Kinesin | KIF2C/ MCAK | +/− * | K | [76] | Ubiquitination | APC/C | + | [73] | |
| KIF10/ CENP-E | + | K | APC3/ CDC27 | + | D | ||||
| KIF11 | − | [73,94] | APC5 | + | D | ||||
| KIF15 | − | [94] | APC6/ CDC16 | + | D | ||||
| KIF18A | + | [95] | APC8/ CDC23 | + | D | ||||
| KIF20A | + | [96] | APC10 | + | D | ||||
| KIF23/ MKLP1 | + | G, L | APC11 | + | D | ||||
| KIF24 | − | [97] | FBX4 | + | L | ||||
| KIFC1/ HSET | + | G, K | [64,74,75] | Polyubiquitin-C | + | L | |||
| Kinetochore | AURKB | + | G, L | USP8 | + | K | |||
| Borealin | + | D, G, L | USP28 | - | [98] | ||||
| BUB1 | + | K | USP31 | + | K | ||||
| CEPN-A | + | K | USP54 | + | L |
6. WGD Induces Genome Instability during Mitosis
7. Size Matters: Promotion of Tumorigenesis after WGD
8. Large Targets: WGD and Cancer Therapies
9. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 2008, 121, 3859–3866. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Nehme, Z. Polyploid Giant Cancer Cells, a Hallmark of Oncoviruses and a New Therapeutic Challenge. Front. Oncol. 2020, 10, 567116. [Google Scholar] [CrossRef] [PubMed]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.Y.C. Cell Cycle Control: A System of Interlinking Oscillators. Methods Mol. Biol. 2021, 2329, 1–18. [Google Scholar] [PubMed]
- Teixeira, L.K.; Reed, S.I. Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 2013, 82, 387–414. [Google Scholar] [CrossRef] [PubMed]
- Haarhuis, J.H.; Elbatsh, A.M.; Rowland, B.D. Cohesin and its regulation: On the logic of X-shaped chromosomes. Dev. Cell 2014, 31, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Sivaprasad, U.; Machida, Y.J.; Dutta, A. APC/C—The master controller of origin licensing. Cell Div. 2007, 2, 8. [Google Scholar] [CrossRef]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Desai, A.; Corbett, K.D. Evolutionary Dynamics and Molecular Mechanisms of HORMA Domain Protein Signaling. Annu. Rev. Biochem. 2022, 91, 541–569. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef]
- Gascoigne, K.E.; Taylor, S.S. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef]
- Wang, Y.; Poon, R.Y.C. MARCH5 regulates mitotic apoptosis through MCL1-dependent and independent mechanisms. Cell Death Differ. 2022. [Google Scholar] [CrossRef]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Lok, T.M.; Wang, Y.; Xu, W.K.; Xie, S.; Ma, H.T.; Poon, R.Y.C. Mitotic slippage is determined by p31comet and the weakening of the spindle-assembly checkpoint. Oncogene 2020, 39, 2819–2834. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.Y. Encyclopedia of Cell Biology; Bradshaw, R.A., Stahl, S.P.D., Eds.; Academic Press: Cambridge, UK, 2016; pp. 399–403. [Google Scholar]
- Shi, Q.; King, R.W. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 2005, 437, 1038–1042. [Google Scholar] [CrossRef]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009, 136, 473–484. [Google Scholar] [CrossRef]
- Cimini, D.; Mattiuzzo, M.; Torosantucci, L.; Degrassi, F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol. Biol. Cell 2003, 14, 3821–3833. [Google Scholar] [CrossRef]
- Gisselsson, D.; Pettersson, L.; Höglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Dal Cin, P.; Mitelman, F.; Mandahl, N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef] [Green Version]
- Cimini, D.; Howell, B.; Maddox, P.; Khodjakov, A.; Degrassi, F.; Salmon, E.D. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 2001, 153, 517–527. [Google Scholar] [CrossRef]
- Yam, C.Q.X.; Lim, H.H.; Surana, U. DNA damage checkpoint execution and the rules of its disengagement. Front. Cell Dev. Biol. 2022, 10, 1020643. [Google Scholar] [CrossRef]
- Chu, K.; Teele, N.; Dewey, M.W.; Albright, N.; Dewey, W.C. Computerized video time lapse study of cell cycle delay and arrest, mitotic catastrophe, apoptosis and clonogenic survival in irradiated 14-3-3sigma and CDKN1A (p21) knockout cell lines. Radiat Res. 2004, 162, 270–286. [Google Scholar] [CrossRef]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef]
- Acilan, C.; Potter, D.M.; Saunders, W.S. DNA repair pathways involved in anaphase bridge formation. Genes Chromosom. Cancer 2007, 46, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Uetake, Y.; Sluder, G. Cell cycle progression after cleavage failure: Mammalian somatic cells do not possess a “tetraploidy checkpoint”. J. Cell Biol. 2004, 165, 609–615. [Google Scholar] [CrossRef]
- Wong, C.; Stearns, T. Mammalian cells lack checkpoints for tetraploidy, aberrant centrosome number, and cytokinesis failure. BMC Cell Biol. 2005, 6, 6. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Hayashi, M.T.; Karlseder, J. pDNA damage associated with mitosis and cytokinesis failure. Oncogene 2013, 32, 4593–4601. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar]
- Woo, R.A.; Poon, R.Y. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2003, 2, 316–324. [Google Scholar] [CrossRef]
- Potapova, T.A.; Seidel, C.W.; Box, A.C.; Rancati, G.; Li, R. Transcriptome analysis of tetraploid cells identifies cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol. Biol. Cell 2016, 27, 3065–3084. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.C.; Corsino, P.E.; Davis, B.J.; Law, M.E.; Nørgaard, P.; Law, B.K. Constitutive Cdk2 activity promotes aneuploidy while altering the spindle assembly and tetraploidy checkpoints. J. Cell Sci. 2013, 126, 1207–1217. [Google Scholar] [CrossRef]
- Senovilla, L.; Vitale, I.; Galluzzi, L.; Vivet, S.; Joza, N.; Younes, A.B.; Rello-Varona, S.; Castedo, M.; Kroemer, G. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle 2009, 8, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Fava, L.L.; Schuler, F.; Sladky, V.; Haschka, M.D.; Soratroi, C.; Eiterer, L.; Demetz, E.; Weiss, G.; Geley, S.; Nigg, E.A.; et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017, 31, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Cornils, H.; Chiu, S.Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Théry, M.; Camargo, F.D.; Pellman, D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Yamamoto, M.; Morita, S.; Kimura, M.; Nagao, Y.; Hatada, I. p53 suppresses tetraploid development in mice. Sci. Rep. 2015, 5, 8907. [Google Scholar] [CrossRef]
- Hayashi, M.T.; Cesare, A.J.; Fitzpatrick, J.A.; Lazzerini-Denchi, E.; Karlseder, J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol. 2012, 19, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Mayer, V.W.; Aguilera, A. High levels of chromosome instability in polyploids of Saccharomyces cerevisiae. Mutat. Res. 1990, 231, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Storchová, Z.; Breneman, A.; Cande, J.; Dunn, J.; Burbank, K.; O’Toole, E.; Pellman, D. Genome-wide genetic analysis of polyploidy in yeast. Nature 2006, 443, 541–547. [Google Scholar] [CrossRef]
- Cowell, J.K.; Wigley, C.B. Changes in DNA content during in vitro transformation of mouse salivary gland epithelium. J. Natl. Cancer Inst. 1980, 64, 1443–1449. [Google Scholar] [CrossRef]
- Ho, C.C.; Hau, P.M.; Marxer, M.; Poon, R.Y. The requirement of p53 for maintaining chromosomal stability during tetraploidization. Oncotarget 2010, 1, 583–595. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Norden, C.; Mendoza, M.; Dobbelaere, J.; Kotwaliwale, C.V.; Biggins, S.; Barral, Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell 2006, 125, 85–98. [Google Scholar] [CrossRef]
- Petsalaki, E.; Zachos, G. The Abscission Checkpoint: A Guardian of Chromosomal Stability. Cells 2021, 10, 3350. [Google Scholar] [CrossRef]
- Pampalona, J.; Frías, C.; Genescà, A.; Tusell, L. Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLoS Genet. 2012, 8, e1002679. [Google Scholar] [CrossRef]
- Pedersen, S.; Karemore, G.; Gudjonsson, T.; Rask, M.B.; Neumann, B.; Hériché, J.K.; Pepperkok, R.; Ellenberg, J.; Gerlich, D.W.; Lukas, J.; et al. Profiling DNA damage response following mitotic perturbations. Nat. Commun. 2016, 7, 13887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemble, S.; Wardenaar, R.; Keuper, K.; Srivastava, N.; Nano, M.; Macé, A.-S.; Tijhuis, A.E.; Bernhard, S.V.; Spierings, D.C.J.; Simon, A.; et al. Genetic instability from a single S phase after whole-genome duplication. Nature 2022, 604, 146–151. [Google Scholar] [CrossRef]
- Wangsa, D.; Quintanilla, I.; Torabi, K.; Vila-Casadesús, M.; Ercilla, A.; Klus, G.; Yuce, Z.; Galofré, C.; Cuatrecasas, M.; Lozano, J.J.; et al. Near-tetraploid cancer cells show chromosome instability triggered by replication stress and exhibit enhanced invasiveness. FASEB J. 2018, 32, 3502–3517. [Google Scholar] [CrossRef]
- Zheng, L.; Dai, H.; Zhou, M.; Li, X.; Liu, C.; Guo, Z.; Wu, X.; Wu, J.; Wang, C.; Zhong, J.; et al. Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nat. Commun. 2012, 3, 815. [Google Scholar] [CrossRef]
- Agircan, F.G.; Schiebel, E.; Mardin, B.R. Separate to operate: Control of centrosome positioning and separation. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130461. [Google Scholar] [CrossRef]
- Gönczy, P.; Hatzopoulos, G.N. Centriole assembly at a glance. J. Cell Sci. 2019, 132, jcs228833. [Google Scholar] [CrossRef]
- Hinchcliffe, E.H.; Li, C.; Thompson, E.A.; Maller, J.L.; Sluder, G. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 1999, 283, 851–854. [Google Scholar] [CrossRef]
- Lacey, K.R.; Jackson, P.K.; Stearns, T. Cyclin-dependent kinase control of centrosome duplication. Proc. Natl. Acad. Sci. USA 1999, 96, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9819–9824. [Google Scholar] [CrossRef]
- Shinmura, K.; Bennett, R.A.; Tarapore, P.; Fukasawa, K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene 2007, 26, 2939–2944. [Google Scholar] [CrossRef]
- Sabat-Pośpiech, D.; Fabian-Kolpanowicz, K.; Prior, I.A.; Coulson, J.M.; Fielding, A.B. Targeting centrosome amplification, an Achilles’ heel of cancer. Biochem. Soc. Trans. 2019, 47, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144. [Google Scholar] [CrossRef] [PubMed]
- Basto, R.; Brunk, K.; Vinadogrova, T.; Peel, N.; Franz, A.; Khodjakov, A.; Raff, J.W. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008, 133, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Sabino, D.; Gogendeau, D.; Gambarotto, D.; Nano, M.; Pennetier, C.; Dingli, F.; Arras, G.; Loew, D.; Basto, R. Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr. Biol. 2015, 25, 879–889. [Google Scholar] [CrossRef]
- Magescas, J.; Zonka, J.C.; Feldman, J.L. A two-step mechanism for the inactivation of microtubule organizing center function at the centrosome. Elife 2019, 8, e47867. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, A.; Soni, K.; Schorpp, K.; Zhao, F.; Minakar, A.; Zheng, X.; Mandad, S.; Macheleidt, I.; Ramani, A.; Kubelka, T.; et al. Inhibition of CPAP-tubulin interaction prevents proliferation of centrosome-amplified cancer cells. EMBO J. 2019, 38, e99876. [Google Scholar] [CrossRef]
- Pannu, V.; Rida, P.C.; Celik, B.; Turaga, R.C.; Ogden, A.; Cantuaria, G.; Gopalakrishnan, J.; Aneja, R. Centrosome-declustering drugs mediate a two-pronged attack on interphase and mitosis in supercentrosomal cancer cells. Cell Death Dis. 2014, 5, e1538. [Google Scholar] [CrossRef]
- Quintyne, N.J.; Reing, J.E.; Hoffelder, D.R.; Gollin, S.M.; Saunders, W.S. Spindle multipolarity is prevented by centrosomal clustering. Science 2005, 307, 127–129. [Google Scholar] [CrossRef]
- Goshima, G.; Wollman, R.; Goodwin, S.S.; Zhang, N.; Scholey, J.M.; Vale, R.D.; Stuurman, N. kGenes required for mitotic spindle assembly in Drosophila S2 cells. Science 2007, 316, 417–421. [Google Scholar] [CrossRef]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef]
- Leber, B.; Maier, B.; Fuchs, F.; Chi, J.; Riffel, P.; Anderhub, S.; Wagner, L.; Ho, A.D.; Salisbury, J.L.; Boutros, M.; et al. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2010, 2, 33ra38. [Google Scholar] [CrossRef] [PubMed]
- Drosopoulos, K.; Tang, C.; Chao, W.C.; Linardopoulos, S. APC/C is an essential regulator of centrosome clustering. Nat. Commun. 2014, 5, 3686. [Google Scholar] [CrossRef]
- Chavali, P.L.; Chandrasekaran, G.; Barr, A.R.; Tátrai, P.; Taylor, C.; Papachristou, E.K.; Woods, C.G.; Chavali, S.; Gergely, F. A CEP215-HSET complex links centrosomes with spindle poles and drives centrosome clustering in cancer. Nat. Commun. 2016, 7, 11005. [Google Scholar] [CrossRef]
- Kleylein-Sohn, J.; Pöllinger, B.; Ohmer, M.; Hofmann, F.; Nigg, E.A.; Hemmings, B.A.; Wartmann, M. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J. Cell Sci. 2012, 125, 5391–5402. [Google Scholar] [CrossRef] [PubMed]
- Goupil, A.; Nano, M.; Letort, G.; Gemble, S.; Edwards, F.; Goundiam, O.; Gogendeau, D.; Pennetier, C.; Basto, R. Chromosomes function as a barrier to mitotic spindle bipolarity in polyploid cells. J. Cell Biol. 2020, 219, e201908006. [Google Scholar] [CrossRef] [PubMed]
- Konotop, G.; Bausch, E.; Nagai, T.; Turchinovich, A.; Becker, N.; Benner, A.; Boutros, M.; Mizuno, K.; Krämer, A.; Raab, M.S. Pharmacological Inhibition of Centrosome Clustering by Slingshot-Mediated Cofilin Activation and Actin Cortex Destabilization. Cancer Res. 2016, 76, 6690–6700. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Bagonis, M.; Danuser, G.; Pellman, D. Direct Microtubule-Binding by Myosin-10 Orients Centrosomes toward Retraction Fibers and Subcortical Actin Clouds. Dev. Cell 2015, 34, 323–337. [Google Scholar] [CrossRef]
- Watanabe, S.; Shioi, G.; Furuta, Y.; Goshima, G. Intra-spindle Microtubule Assembly Regulates Clustering of Microtubule-Organizing Centers during Early Mouse Development. Cell Rep. 2016, 15, 54–60. [Google Scholar] [CrossRef]
- Navarro-Serer, B.; Childers, E.P.; Hermance, N.M.; Mercadante, D.; Manning, A.L. Aurora A inhibition limits centrosome clustering and promotes mitotic catastrophe in cells with supernumerary centrosomes. Oncotarget 2019, 10, 1649–1659. [Google Scholar] [CrossRef]
- Fielding, A.B.; Lim, S.; Montgomery, K.; Dobreva, I.; Dedhar, S. A critical role of integrin-linked kinase, ch-TOG and TACC3 in centrosome clustering in cancer cells. Oncogene 2011, 30, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Lu, Y.; Orr, B.; Godek, K.; Mustachio, L.M.; Kawakami, M.; Sekula, D.; Compton, D.A.; Freemantle, S.; Dmitrovsky, E. Specific CP110 Phosphorylation Sites Mediate Anaphase Catastrophe after CDK2 Inhibition: Evidence for Cooperation with USP33 Knockdown. Mol. Cancer 2015, 14, 2576–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuer, M.; Kolano, A.; Kwon, M.; Li, C.C.; Tsai, T.F.; Pellman, D.; Brunet, S.; Verlhac, M.H. HURP permits MTOC sorting for robust meiotic spindle bipolarity, similar to extra centrosome clustering in cancer cells. J. Cell Biol. 2010, 191, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.; O’Regan, L.; Dyer, M.J.S.; Bayliss, R.; Fry, A.M. Hsp72 and Nek6 Cooperate to Cluster Amplified Centrosomes in Cancer Cells. Cancer Res. 2017, 77, 4785–4796. [Google Scholar] [CrossRef] [PubMed]
- Vitre, B.; Taulet, N.; Guesdon, A.; Douanier, A.; Dosdane, A.; Cisneros, M.; Maurin, J.; Hettinger, S.; Anguille, C.; Taschner, M.; et al. IFT proteins interact with HSET to promote supernumerary centrosome clustering in mitosis. EMBO Rep. 2020, 21, e49234. [Google Scholar] [CrossRef]
- Rhys, A.D.; Monteiro, P.; Smith, C.; Vaghela, M.; Arnandis, T.; Kato, T.; Leitinger, B.; Sahai, E.; McAinsh, A.; Charras, G.; et al. Loss of E-cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J. Cell Biol. 2018, 217, 195–209. [Google Scholar] [CrossRef]
- Tilwani, S.; Gandhi, K.; Narayan, S.; Ainavarapu, S.R.K.; Dalal, S.N. Disruption of desmosome function leads to increased centrosome clustering in 14-3-3γ-knockout cells with supernumerary centrosomes. FEBS Lett. 2021, 595, 2675–2690. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Mustachio, L.M.; Liu, X.; Dmitrovsky, E. Engaging Anaphase Catastrophe Mechanisms to Eradicate Aneuploid Cancers. Mol. Cancer 2018, 17, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Sun, L.; Meng, L.; Hu, C.; Wang, X.; Shi, Z.; Hu, C.; Han, Y.; Yang, Q.; Cao, L.; et al. The ATM and ATR kinases regulate centrosome clustering and tumor recurrence by targeting KIFC1 phosphorylation. Nat. Commun. 2021, 12, 20. [Google Scholar] [CrossRef]
- Yi, Q.; Zhao, X.; Huang, Y.; Ma, T.; Zhang, Y.; Hou, H.; Cooke, H.J.; Yang, D.Q.; Wu, M.; Shi, Q. p53 dependent centrosome clustering prevents multipolar mitosis in tetraploid cells. PLoS ONE 2011, 6, e27304. [Google Scholar] [CrossRef]
- Johannes, J.W.; Almeida, L.; Daly, K.; Ferguson, A.D.; Grosskurth, S.E.; Guan, H.; Howard, T.; Ioannidis, S.; Kazmirski, S.; Lamb, M.L.; et al. Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorg. Med. Chem. Lett. 2015, 25, 5743–5747. [Google Scholar] [CrossRef]
- Antao, N.V.; Marcet-Ortega, M.; Cifani, P.; Kentsis, A.; Foley, E.A. A Cancer-Associated Missense Mutation in PP2A-Aα Increases Centrosome Clustering during Mitosis. iScience 2019, 19, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, E.J.; Kawamura, E.; Gillespie, J.A.; Balgi, A.; Kannan, N.; Muller, W.J.; Roberge, M.; Dedhar, S. Stat3 regulates centrosome clustering in cancer cells via Stathmin/PLK1. Nat. Commun. 2017, 8, 15289. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Iimori, M.; Wakasa, T.; Ando, K.; Saeki, H.; Oda, Y.; Oki, E.; Maehara, Y. The balance of forces generated by kinesins controls spindle polarity and chromosomal heterogeneity in tetraploid cells. J. Cell Sci. 2019, 132, jcs231530. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef]
- Xie, B.; Pu, Y.; Yang, F.; Chen, W.; Yue, W.; Ma, J.; Zhang, N.; Jiang, Y.; Wu, J.; Lin, Y.; et al. Proteomic Mapping and Targeting of Mitotic Pericentriolar Material in Tumors Bearing Centrosome Amplification. Cancer Res. 2022, 82, 2576–2592. [Google Scholar] [CrossRef]
- Mashima, Y.; Nohira, H.; Sugihara, H.; Dynlacht, B.D.; Kobayashi, T.; Itoh, H. KIF24 depletion induces clustering of supernumerary centrosomes in PDAC cells. Life Sci. Alliance 2022, 5, e202201470. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, S.V.; Seget-Trzensiok, K.; Kuffer, C.; Krastev, D.B.; Stautmeister, L.M.; Theis, M.; Keuper, K.; Boekenkamp, J.E.; Kschischo, M.; Buchholz, F.; et al. Loss of USP28 and SPINT2 expression promotes cancer cell survival after whole genome doubling. Cell. Oncol. 2022, 45, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Silkworth, W.T.; Nardi, I.K.; Scholl, L.M.; Cimini, D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS ONE 2009, 4, e6564. [Google Scholar] [CrossRef]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffelder, D.R.; Luo, L.; Burke, N.A.; Watkins, S.C.; Gollin, S.M.; Saunders, W.S. Resolution of anaphase bridges in cancer cells. Chromosoma 2004, 112, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Nano, M.; Gemble, S.; Simon, A.; Pennetier, C.; Fraisier, V.; Marthiens, V.; Basto, R. Cell-Cycle Asynchrony Generates DNA Damage at Mitotic Entry in Polyploid Cells. Curr. Biol. 2019, 29, 3937–3945.e7. [Google Scholar] [CrossRef]
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Orr-Weaver, T.L. When bigger is better: The role of polyploidy in organogenesis. Trends Genet. 2015, 31, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, J.; Ravid, K. Polyploidy: Occurrence in nature, mechanisms, and significance for the megakaryocyte-platelet system. Exp. Hematol. 2000, 28, 3–16. [Google Scholar] [CrossRef]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: New York, NY, USA, 1970. [Google Scholar]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Hammarlund, E.U.; Austin, R.H.; Axelrod, R.; Brown, J.S.; Amend, S.R. Cancer cells employ an evolutionarily conserved polyploidization program to resist therapy. In Seminars in Cancer Biology; Academic Press: Cambridge, UK, 2020. [Google Scholar]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Grönroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef]
- López, S.; Lim, E.L.; Horswell, S.; Haase, K.; Huebner, A.; Dietzen, M.; Mourikis, T.P.; Watkins, T.B.K.; Rowan, A.; Dewhurst, S.M.; et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 2020, 52, 283–293. [Google Scholar] [CrossRef]
- Barrett, M.T.; Pritchard, D.; Palanca-Wessels, C.; Anderson, J.; Reid, B.J.; Rabinovitch, P.S. Molecular phenotype of spontaneously arising 4N (G2-tetraploid) intermediates of neoplastic progression in Barrett’s esophagus. Cancer Res. 2003, 63, 4211–4217. [Google Scholar] [PubMed]
- Galipeau, P.C.; Cowan, D.S.; Sanchez, C.A.; Barrett, M.T.; Emond, M.J.; Levine, D.S.; Rabinovitch, P.S.; Reid, B.J. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proc. Natl. Acad. Sci. USA 1996, 93, 7081–7084. [Google Scholar] [CrossRef] [PubMed]
- Olaharski, A.J.; Sotelo, R.; Solorza-Luna, G.; Gonsebatt, M.E.; Guzman, P.; Mohar, A.; Eastmond, D.A. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 2006, 27, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Senovilla, L.; Criollo, A.; Jemaà, M.; Castedo, M.; Kroemer, G. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2011, 18, 1403–1413. [Google Scholar] [CrossRef]
- Duelli, D.M.; Padilla-Nash, H.M.; Berman, D.; Murphy, K.M.; Ried, T.; Lazebnik, Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 2007, 17, 431–437. [Google Scholar] [CrossRef]
- Al-Rawi, D.H.; Bakhoum, S.F. Chromosomal instability as a source of genomic plasticity. Curr. Opin. Genet. Dev. 2022, 74, 101913. [Google Scholar] [CrossRef]
- King, R.W. When 2 + 2 = 5: The origins and fates of aneuploid and tetraploid cells. Biochim. Biophys. Acta 2008, 1786, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Harding, A. Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qiao, Q.; Xu, H.; Zhou, R.; Liu, X. Human cell polyploidization: The good and the evil. Semin. Cancer Biol. 2022, 81, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Hau, P.M.; Siu, W.Y.; Wong, N.; Lai, P.B.; Poon, R.Y. Polyploidization increases the sensitivity to DNA-damaging agents in mammalian cells. FEBS Lett. 2006, 580, 4727–4736. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.H.; Kim, J.; Ahn, J.; Hwang, S.G.; Oh, J.S.; Kim, J.S. Centrosome Clustering Is a Tumor-selective Target for the Improvement of Radiotherapy in Breast Cancer Cells. Anticancer Res. 2018, 38, 3393–3400. [Google Scholar] [CrossRef] [PubMed]
- Watts, C.A.; Richards, F.M.; Bender, A.; Bond, P.J.; Korb, O.; Kern, O.; Riddick, M.; Owen, P.; Myers, R.M.; Raff, J.; et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem. Biol. 2013, 20, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mikule, K.; Wang, W.; Su, N.; Petteruti, P.; Gharahdaghi, F.; Code, E.; Zhu, X.; Jacques, K.; Lai, Z.; et al. Discovery and mechanistic study of a small molecule inhibitor for motor protein KIFC1. ACS Chem. Biol. 2013, 8, 2201–2208. [Google Scholar] [CrossRef]
- Czechanski, A.; Kim, H.; Byers, C.; Greenstein, I.; Stumpff, J.; Reinholdt, L.G. Kif18a is specifically required for mitotic progression during germ line development. Dev. Biol. 2015, 402, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Zhao, X.D.; Wang, X.; Yao, Y.X.; Zhang, L.L.; Shu, R.Z.; Ren, W.H.; Huang, Y.; Huang, L.; Gu, M.M.; et al. Germinal Cell Aplasia in Kif18a Mutant Male Mice due to Impaired Chromosome Congression and Dysregulated BubR1 and CENP-E. Genes Cancer 2010, 1, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Marquis, C.; Fonseca, C.L.; Queen, K.A.; Wood, L.; Vandal, S.E.; Malaby, H.L.H.; Clayton, J.E.; Stumpff, J. Chromosomally unstable tumor cells specifically require KIF18A for proliferation. Nat. Commun. 2021, 12, 1213. [Google Scholar] [CrossRef] [PubMed]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.T.; Gall, J.G.; Spradling, A.C. Error-prone polyploid mitosis during normal Drosophila development. Genes Dev. 2010, 24, 2294–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, T.Y.; Poon, R.Y.C. Whole-Genome Duplication and Genome Instability in Cancer Cells: Double the Trouble. Int. J. Mol. Sci. 2023, 24, 3733. https://doi.org/10.3390/ijms24043733
Lau TY, Poon RYC. Whole-Genome Duplication and Genome Instability in Cancer Cells: Double the Trouble. International Journal of Molecular Sciences. 2023; 24(4):3733. https://doi.org/10.3390/ijms24043733
Chicago/Turabian StyleLau, Tsz Yin, and Randy Y.C. Poon. 2023. "Whole-Genome Duplication and Genome Instability in Cancer Cells: Double the Trouble" International Journal of Molecular Sciences 24, no. 4: 3733. https://doi.org/10.3390/ijms24043733