

Reducing GEF-H1 Expression Inhibits Renal Cyst Formation, Inflammation, and Fibrosis via RhoA Signaling in Nephronophthisis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
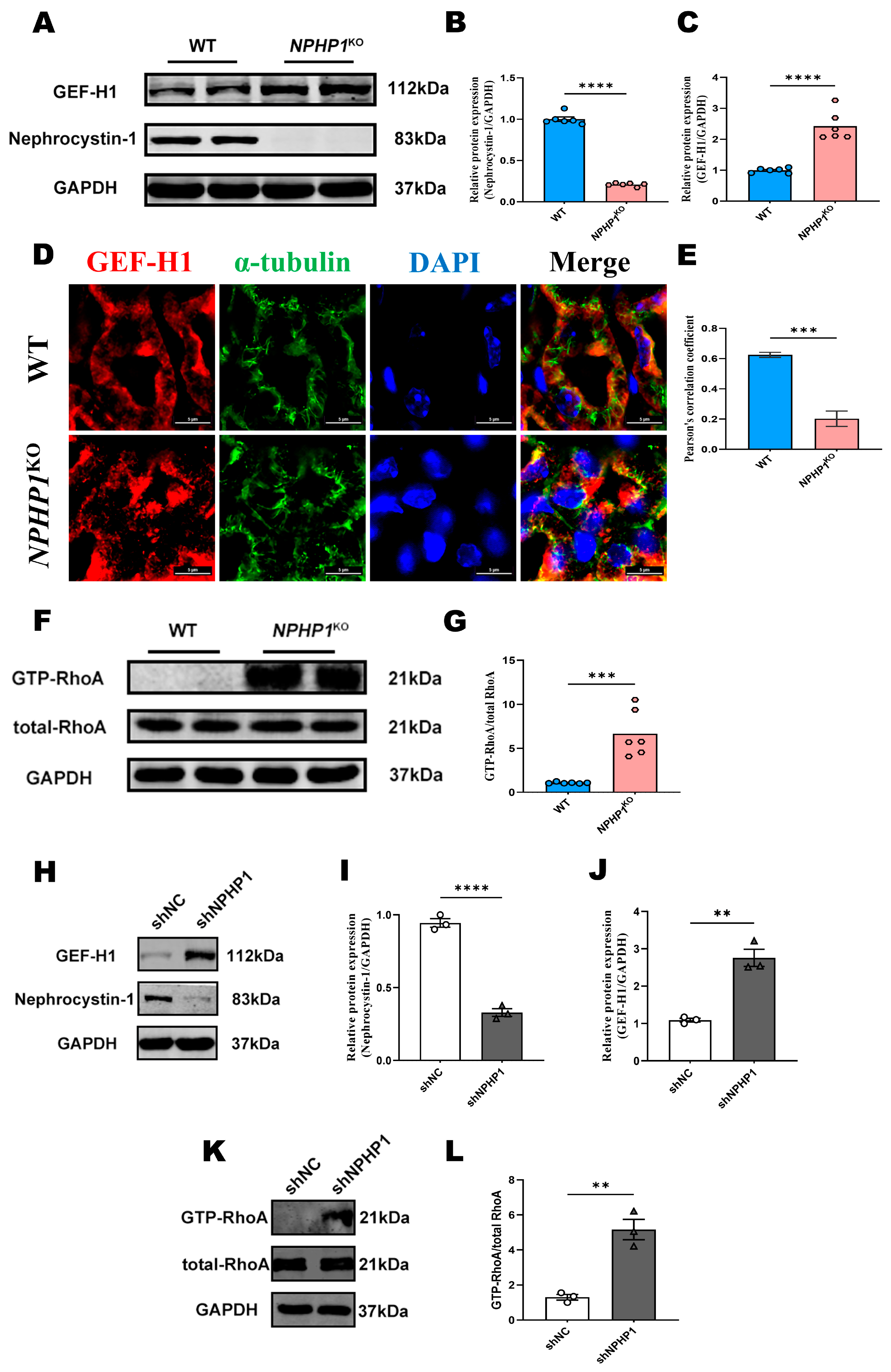
2.1. The GEF-H1/RhoA Pathway Was Activated in the Renal Tissue of NPHP1KO Mice and NPHP1KD HK2 Cells
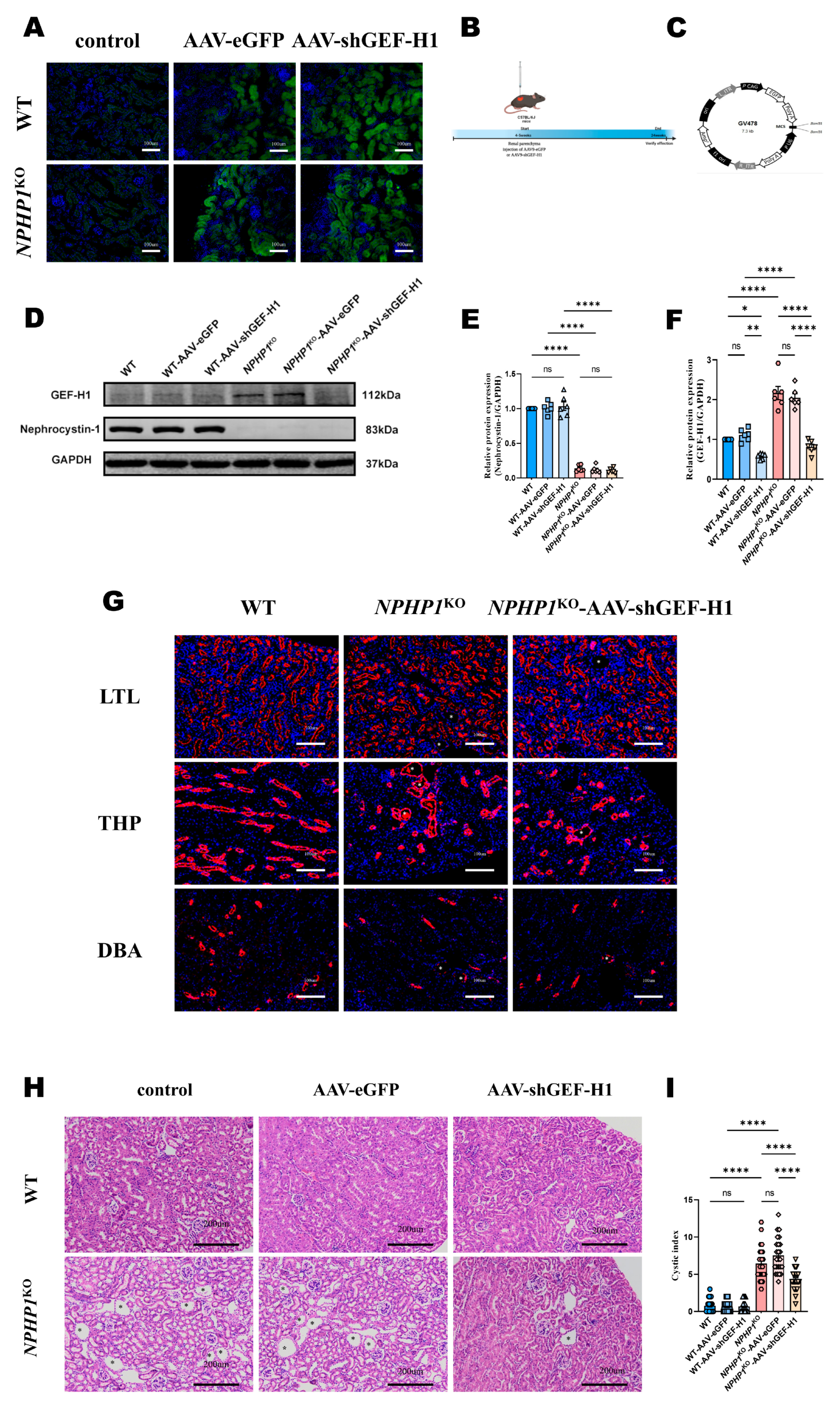
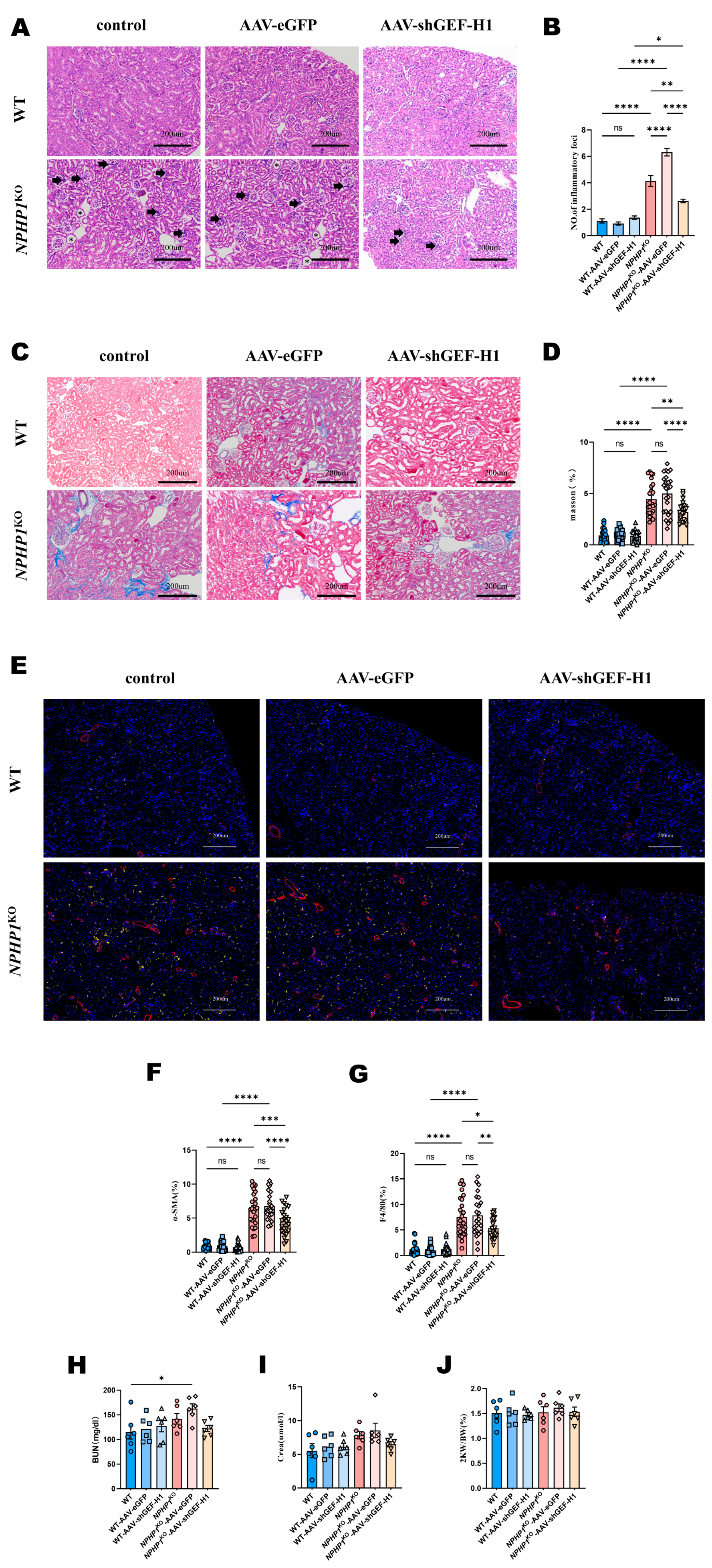
2.2. Knockdown of GEF-H1 Alleviated Renal Histological Injuries in NPHP1 KO Mice
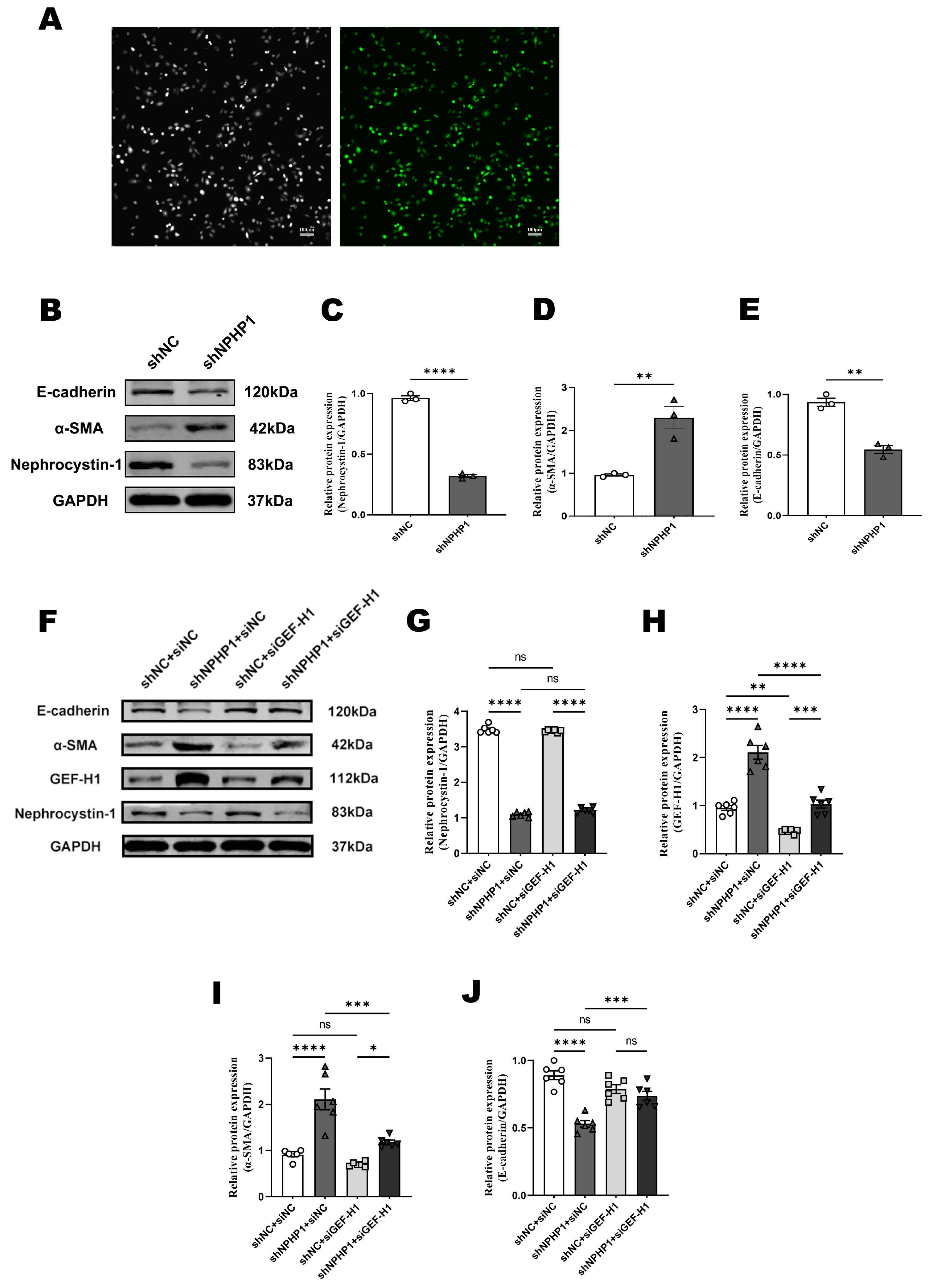
2.3. GEF-H1 Knockdown Alleviated Epithelial-Mesenchymal Transition (EMT) in NPHP1KD HK2 Cells
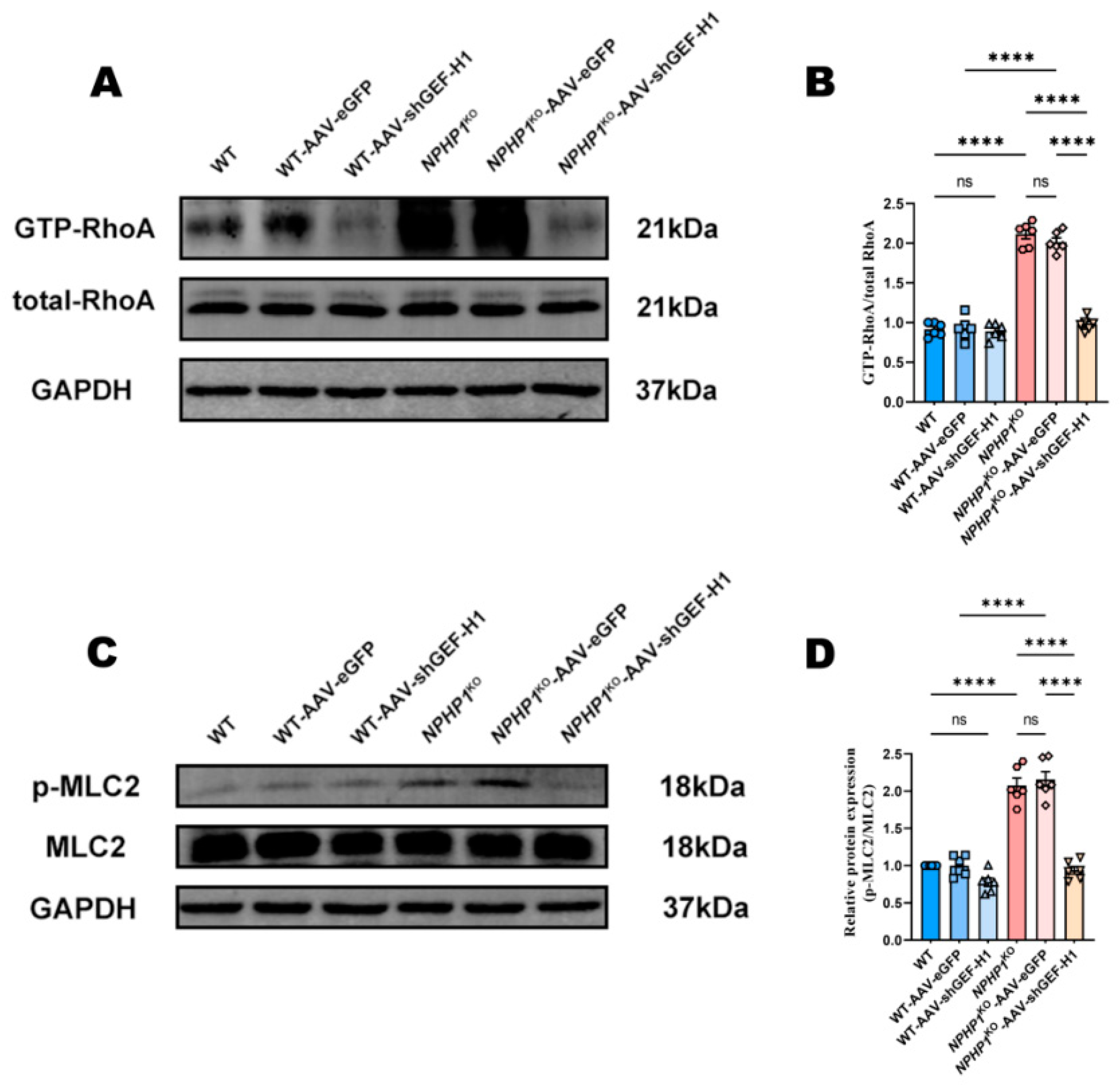
2.4. The GEF-H1/RhoA Pathway Was Associated with MLC2 Phosphorylation in NPHP1KO Mice
3. Discussion
4. Materials and Methods
4.1. Mice and Antibodies
4.2. AAV9 Injection in Situ
4.3. Urea and Creatinine Measurements
4.4. Cell Culture
4.5. Lentiviral Transfection
4.6. Transfection of siRNA
4.7. Protein Extraction and Western Blotting
4.8. RhoA GTPase Activation Assay
4.9. Immunofluorescence
4.10. Histopathological Analysis
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hildebrandt, F.; Attanasio, M.; Otto, E. Nephronophthisis: Disease Mechanisms of a Ciliopathy. J. Am. Soc. Nephrol. 2009, 20, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Molinari, E.; Raman, S.; Sayer, J.A. Many Genes—One Disease? Genetics of Nephronophthisis (NPHP) and NPHP-Associated Disorders. Front. Pediatr. 2018, 5, 287. [Google Scholar] [CrossRef]
- Luo, F.; Tao, Y.H. Nephronophthisis: A review of genotype–phenotype correlation. Nephrology 2018, 23, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Simms, R.J.; Eley, L.; Sayer, J.A. Nephronophthisis. Eur. J. Hum. Genet. 2009, 17, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Halbritter, J.; Porath, J.D.; Diaz, K.A.; Braun, D.A.; Kohl, S.; Chaki, M.; Allen, S.J.; Soliman, N.A.; Hildebrandt, F.; Otto, E.A.; et al. Identification of 99 novel mutations in a worldwide cohort of 1056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 2013, 132, 865–884. [Google Scholar] [CrossRef] [PubMed]
- Konrad, M.; Saunier, S.; Calado, J.; Gubler, M.-C.; Broyer, M.; Antignac, C. Familial juvenile nephronophthisis. J. Mol. Med. 1998, 76, 310–316. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Rensing, C.; Betz, R.C.; Sommer, U.; Birnbaum, S.; Imm, A.; Omran, H.; Leipoldt, M.; Otto, E.; The Arbeitsgemeinschaft Für Paediatrische Nephrologie (APN) Study Group; et al. Establishing an algorithm for molecular genetic diagnostics in 127 families with juvenile nephronophthisis. Kidney Int. 2001, 59, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Saunier, S.; Calado, J.; Benessy, F.; Silbermann, F.; Heilig, R.; Weissenbach, J.; Antignac, C. Characterization of the NPHP1 Locus: Mutational Mechanism Involved in Deletions in Familial Juvenile Nephronophthisis. Am. J. Hum. Genet. 2000, 66, 778–789. [Google Scholar] [CrossRef]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 2021, 77, 410–419. [Google Scholar] [CrossRef]
- Waldherr, R.; Lennert, T.; Weber, H.-P.; Födisch, H.J.; Schärer, K. The nephronophthisis complex: A clinicopathologic study in children. Virchows Arch. A 1982, 394, 235–254. [Google Scholar] [CrossRef]
- Srivastava, S.; Sayer, J.A. Nephronophthisis. J. Pediatr. Genet. 2014, 3, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.T. Nephronophthisis and related syndromes. Curr. Opin. Pediatr. 2015, 27, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.W.; Hildebrandt, F. Mechanisms of Nephronophthisis and Related Ciliopathies. Nephron Exp. Nephrol. 2011, 118, e9–e14. [Google Scholar] [CrossRef]
- Salomon, R.; Saunier, S.; Niaudet, P. Nephronophthisis. Pediatr. Nephrol. 2009, 24, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2016, 9, a028191. [Google Scholar] [CrossRef]
- Streets, A.J.; Prosseda, P.P.; Ong, A.C. Polycystin-1 regulates ARHGAP35-dependent centrosomal RhoA activation and ROCK signaling. JCI Insight. 2020, 5, e135385. [Google Scholar] [CrossRef]
- Garcia, H.; Serafin, A.S.; Silbermann, F.; Porée, E.; Viau, A.; Mahaut, C.; Billot, K.; Birgy, É.; Garfa-Traore, M.; Roy, S.; et al. Agonists of prostaglandin E 2 receptors as potential first in class treatment for nephronophthisis and related ciliopathies. Proc. Natl. Acad. Sci. USA 2022, 119, e2115960119. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef]
- Birukova, A.A.; Adyshev, D.; Gorshkov, B.; Bokoch, G.M.; Birukov, K.G.; Verin, A.D. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L540–L548. [Google Scholar] [CrossRef] [Green Version]
- Numaga-Tomita, T.; Kitajima, N.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; Birnbaumer, L.; et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 2016, 6, 39383. [Google Scholar] [CrossRef] [PubMed]
- Fine, N.; Dimitriou, I.D.; Rullo, J.; Sandí, M.J.; Petri, B.; Haitsma, J.; Ibrahim, H.; La Rose, J.; Glogauer, M.; Kubes, P.; et al. GEF-H1 is necessary for neutrophil shear stress–induced migration during inflammation. J. Cell Biol. 2016, 215, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, J.; Yang, J.; Yang, X.; He, J.; Wang, R.; Liu, S.; Zhou, L.; Ma, L. Guanine nucleotide exchange factor -H1 promotes inflammatory cytokine production and intracellular mycobacterial elimination in macrophages. Cell Cycle 2017, 16, 1695–1704. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, H.; Zhou, S.; Meng, B.; Liu, Z.-J.; Ouyang, S. Crystal structure of hGEF-H1 PH domain provides insight into incapability in phosphoinositide binding. Biochem. Biophys. Res. Commun. 2016, 471, 621–627. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Lee, H.-H.; Chen, Y.-J.; Bokoch, G.M.; Chang, Z.-F. Contribution of guanine exchange factor H1 in phorbol ester-induced apoptosis. Cell Death Differ. 2006, 13, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Callow, M.G.; Zozulya, S.; Gishizky, M.L.; Jallal, B.; Smeal, T. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 2005, 118, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Zenke, F.T.; Krendel, M.; DerMardirossian, C.; King, C.C.; Bohl, B.P.; Bokoch, G.M. p21-activated Kinase 1 Phosphorylates and Regulates 14-3-3 Binding to GEF-H1, a Microtubule-localized Rho Exchange Factor. J. Biol. Chem. 2004, 279, 18392–18400. [Google Scholar] [CrossRef]
- Azoitei, M.L.; Noh, J.; Marston, D.J.; Roudot, P.; Marshall, C.B.; Daugird, T.A.; Lisanza, S.L.; Sandí, M.-J.; Ikura, M.; Sondek, J.; et al. Spatiotemporal dynamics of GEF-H1 activation controlled by microtubule- and Src-mediated pathways. J. Cell Biol. 2019, 218, 3077–3097. [Google Scholar] [CrossRef]
- Meiri, D.; Marshall, C.B.; Greeve, M.A.; Kim, B.; Balan, M.; Suarez, F.; Bakal, C.; Wu, C.; LaRose, J.; Fine, N.; et al. Mechanistic Insight into the Microtubule and Actin Cytoskeleton Coupling through Dynein-Dependent RhoGEF Inhibition. Mol. Cell 2012, 45, 642–655. [Google Scholar] [CrossRef]
- Sandí, M.-J.; Marshall, C.B.; Balan, M.; Coyaud, É.; Zhou, M.; Monson, D.M.; Ishiyama, N.; Chandrakumar, A.A.; La Rose, J.; Couzens, A.L.; et al. MARK3-mediated phosphorylation of ARHGEF2 couples microtubules to the actin cytoskeleton to establish cell polarity. Sci. Signal. 2017, 10, eaan3286. [Google Scholar] [CrossRef] [Green Version]
- Mollet, G.; Silbermann, F.; Delous, M.; Salomon, R.; Antignac, C.; Saunier, S. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum. Mol. Genet. 2005, 14, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Delous, M.; Hellman, N.E.; Gaudé, H.-M.; Silbermann, F.; Le Bivic, A.; Salomon, R.; Antignac, C.; Saunier, S. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum. Mol. Genet. 2009, 18, 4711–4723. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.C.; Dise, R.S.; Ritchie, M.D.; Hanks, S.K. Nephrocystin-conserved Domains Involved in Targeting to Epithelial Cell-Cell Junctions, Interaction with Filamins, and Establishing Cell Polarity. J. Biol. Chem. 2002, 277, 29028–29035. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.; Aijaz, S.; Leefa Chong San, I.V.; Balda, M.S.; Matter, K. The Y-box factor ZONAB/DbpA associates with GEF-H1/Lfc and mediates Rho-stimulated transcription. EMBO Rep. 2009, 10, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Sun, L.; Liu, L.; Chen, H.; Wang, H.; Li, X.; Liu, T.; Yue, Z. NPHP1 knockdown induces epithelial-mesenchymal transition in Madin-Darby canine kidney cells. Chin. J. Nephrol. 2017, 33, 781–787. [Google Scholar] [CrossRef]
- Shao, F.; Han, D.; Shen, Y.; Bian, W.; Zou, L.; Hu, Y.; Sun, W. Oxycodone relieves permeability damage and apoptosis of oxygen-glucose deprivation/reoxygenation-induced brain microvascular endothelial cells through ras homolog family member A (RhoA)/ Rho-associated coiled-coil containing kinases (ROCK)/myosin light chain 2 (MLC2) signal. Bioengineered 2022, 13, 5205–5215. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.E.L.; Lake, A.V.R.; Johnson, C.A. Primary Cilia, Ciliogenesis and the Actin Cytoskeleton: A Little Less Resorption, a Little More Actin Please. Front. Cell Dev. Biol. 2020, 8, 622822. [Google Scholar] [CrossRef]
- Siesser, P.F.; Motolese, M.; Walker, M.P.; Goldfarb, D.; Gewain, K.; Yan, F.; Kulikauskas, R.M.; Chien, A.J.; Wordeman, L.; Major, M.B. FAM123A Binds to Microtubules and Inhibits the Guanine Nucleotide Exchange Factor ARHGEF2 to Decrease Actomyosin Contractility. Sci. Signal. 2012, 5, ra64. [Google Scholar] [CrossRef]
- Otto, E.A.; Schermer, B.; Obara, T.; O’Toole, J.F.; Hiller, K.S.; Mueller, A.M.; Ruf, R.G.; Hoefele, J.; Beekmann, F.; Landau, D.; et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003, 34, 413–420. [Google Scholar] [CrossRef]
- Togawa, H.; Nakanishi, K.; Mukaiyama, H.; Hama, T.; Shima, Y.; Sako, M.; Miyajima, M.; Nozu, K.; Nishii, K.; Nagao, S.; et al. Epithelial-to-mesenchymal transition in cyst lining epithelial cells in an orthologous PCK rat model of autosomal-recessive polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2011, 300, F511–F520. [Google Scholar] [CrossRef] [Green Version]
- Attanasio, M.; Uhlenhaut, N.H.; Sousa, V.H.; O’Toole, J.F.; Otto, E.; Anlag, K.; Klugmann, C.; Treier, A.-C.; Helou, J.; Sayer, J.A.; et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007, 39, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Schieren, G.; Rumberger, B.; Klein, M.; Kreutz, C.; Wilpert, J.; Geyer, M.; Faller, D.; Timmer, J.; Quack, I.; Rump, L.C.; et al. Gene profiling of polycystic kidneys. Nephrol. Dial. Transplant. 2006, 21, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Shaye, D.D.; Soto, M.C. Epithelial morphogenesis, tubulogenesis and forces in organogenesis. Curr. Top. Dev. Biol. 2021, 144, 161–214. [Google Scholar] [CrossRef] [PubMed]
- Sigurbjörnsdóttir, S.; Mathew, R.; Leptin, M. Molecular mechanisms of de novo lumen formation. Nat. Rev. Mol. Cell Biol. 2014, 15, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Camelo, C.; Luschnig, S. Cells into tubes: Molecular and physical principles underlying lumen formation in tubular organs. Curr. Top. Dev. Biol. 2021, 143, 37–74. [Google Scholar] [CrossRef]
- Wu, X.; Wang, H.; Chen, H.; Lin, H.; Li, M.; Yue, Z.; Sun, L. Overexpression of smad7 inhibits the TGF-β/Smad signaling pathway and EMT in NPHP1-defective MDCK cells. Biochem. Biophys. Res. Commun. 2021, 582, 57–63. [Google Scholar] [CrossRef]
- Mills, C.; Hemkemeyer, S.A.; Alimajstorovic, Z.; Bowers, C.; Eskandarpour, M.; Greenwood, J.; Calder, V.; Chan, A.W.E.; Gane, P.J.; Selwood, D.L.; et al. Therapeutic Validation of GEF-H1 Using a De Novo Designed Inhibitor in Models of Retinal Disease. Cells 2022, 11, 1733. [Google Scholar] [CrossRef]
- Tsapara, A.; Luthert, P.; Greenwood, J.; Hill, C.S.; Matter, K.; Balda, M.S. The RhoA Activator GEF-H1/Lfc Is a Transforming Growth Factor-β Target Gene and Effector That Regulates α-Smooth Muscle Actin Expression and Cell Migration. Mol. Biol. Cell 2010, 21, 860–870. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Kuwae, A.; Yoshida, S.; Sasakawa, C.; Abe, A. Enteropathogenic Escherichia coli activates the RhoA signaling pathway via the stimulation of GEF-H1. EMBO J. 2004, 23, 3570–3582. [Google Scholar] [CrossRef]
- Hiyoshi, H.; Okada, R.; Matsuda, S.; Gotoh, K.; Akeda, Y.; Iida, T.; Kodama, T. Interaction between the Type III Effector VopO and GEF-H1 Activates the RhoA-ROCK Pathway. PLoS Pathog. 2015, 11, e1004694. [Google Scholar] [CrossRef]
- Guo, F.; Xing, Y.; Zhou, Z.; Dou, Y.; Tang, J.; Gao, C.; Huan, J. Guanine-Nucleotide Exchange Factor H1 Mediates Lipopolysaccharide-Induced Interleukin 6 and Tumor Necrosis Factor α Expression in Endothelial Cells Via Activation of Nuclear Factor κB. Shock 2012, 37, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hu, M.; Chen, H.; Wu, X.; Wei, X.; Lin, H.; Gao, X.; Wang, H.; Li, M.; Ong, A.C.M.; et al. An Nphp1 knockout mouse model targeting exon 2–20 demonstrates characteristic phenotypes of human nephronophthisis. Hum. Mol. Genet. 2021, 31, 232–243. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Q.; Lai, J.; Chen, H.; Cai, Y.; Yue, Z.; Lin, H.; Sun, L. Reducing GEF-H1 Expression Inhibits Renal Cyst Formation, Inflammation, and Fibrosis via RhoA Signaling in Nephronophthisis. Int. J. Mol. Sci. 2023, 24, 3504. https://doi.org/10.3390/ijms24043504
Hu Q, Lai J, Chen H, Cai Y, Yue Z, Lin H, Sun L. Reducing GEF-H1 Expression Inhibits Renal Cyst Formation, Inflammation, and Fibrosis via RhoA Signaling in Nephronophthisis. International Journal of Molecular Sciences. 2023; 24(4):3504. https://doi.org/10.3390/ijms24043504
Chicago/Turabian StyleHu, Qiulei, Jiayong Lai, Huamu Chen, Yong Cai, Zhihui Yue, Hongrong Lin, and Liangzhong Sun. 2023. "Reducing GEF-H1 Expression Inhibits Renal Cyst Formation, Inflammation, and Fibrosis via RhoA Signaling in Nephronophthisis" International Journal of Molecular Sciences 24, no. 4: 3504. https://doi.org/10.3390/ijms24043504