


Rescue of Rare CFTR Trafficking Mutants Highlights a Structural Location-Dependent Pattern for Correction


Abstract
:1. Introduction
2. Results
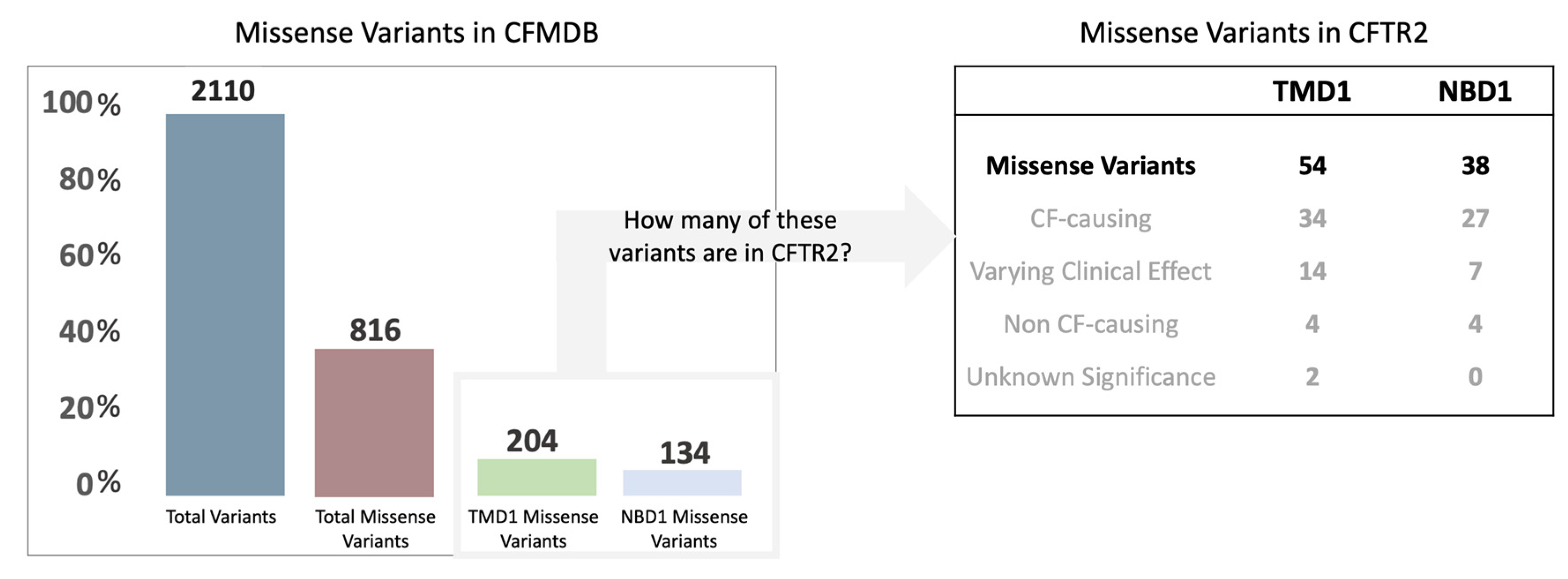
2.1. Rationale and Context for the Mutations Analyzed
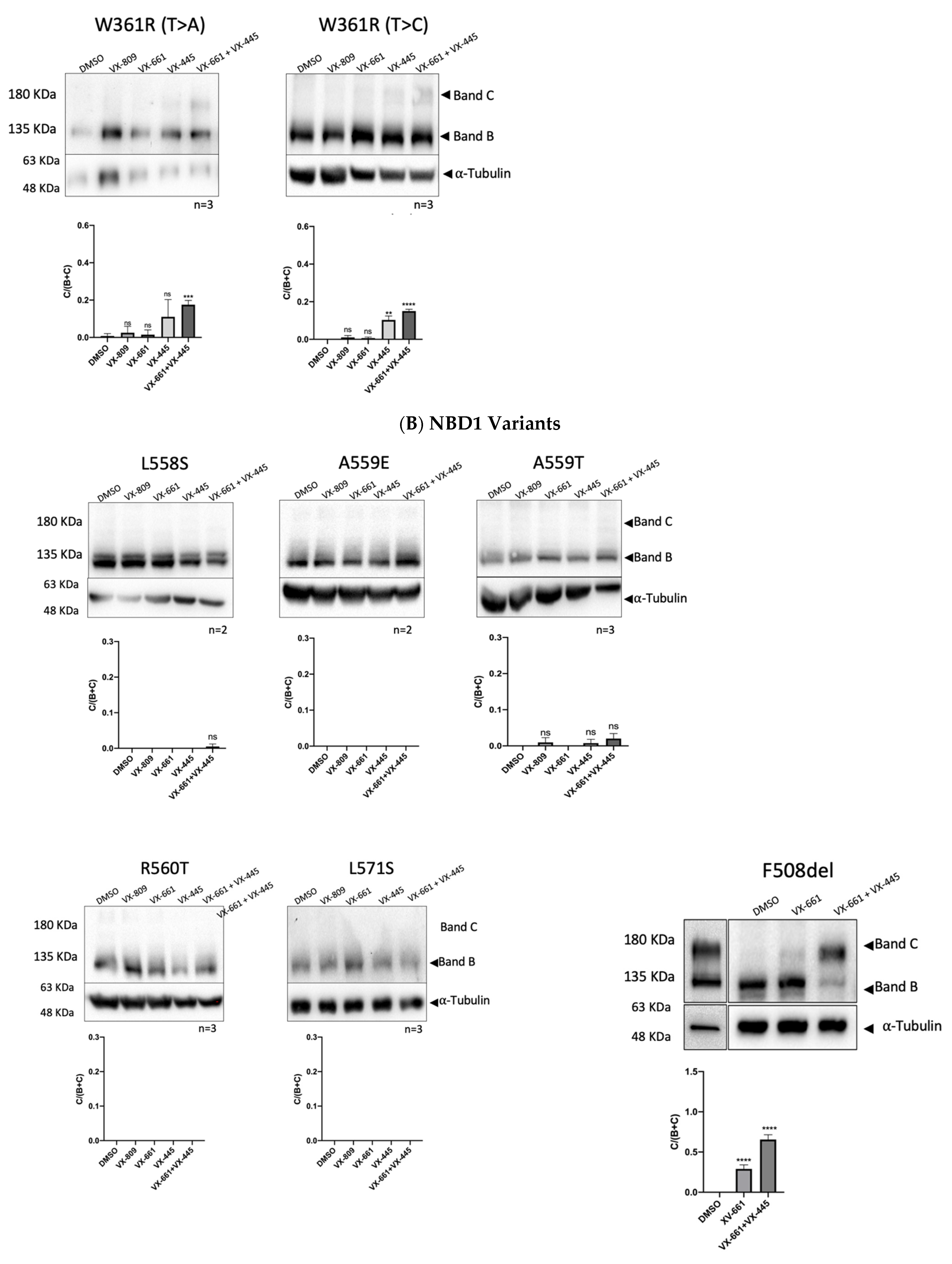
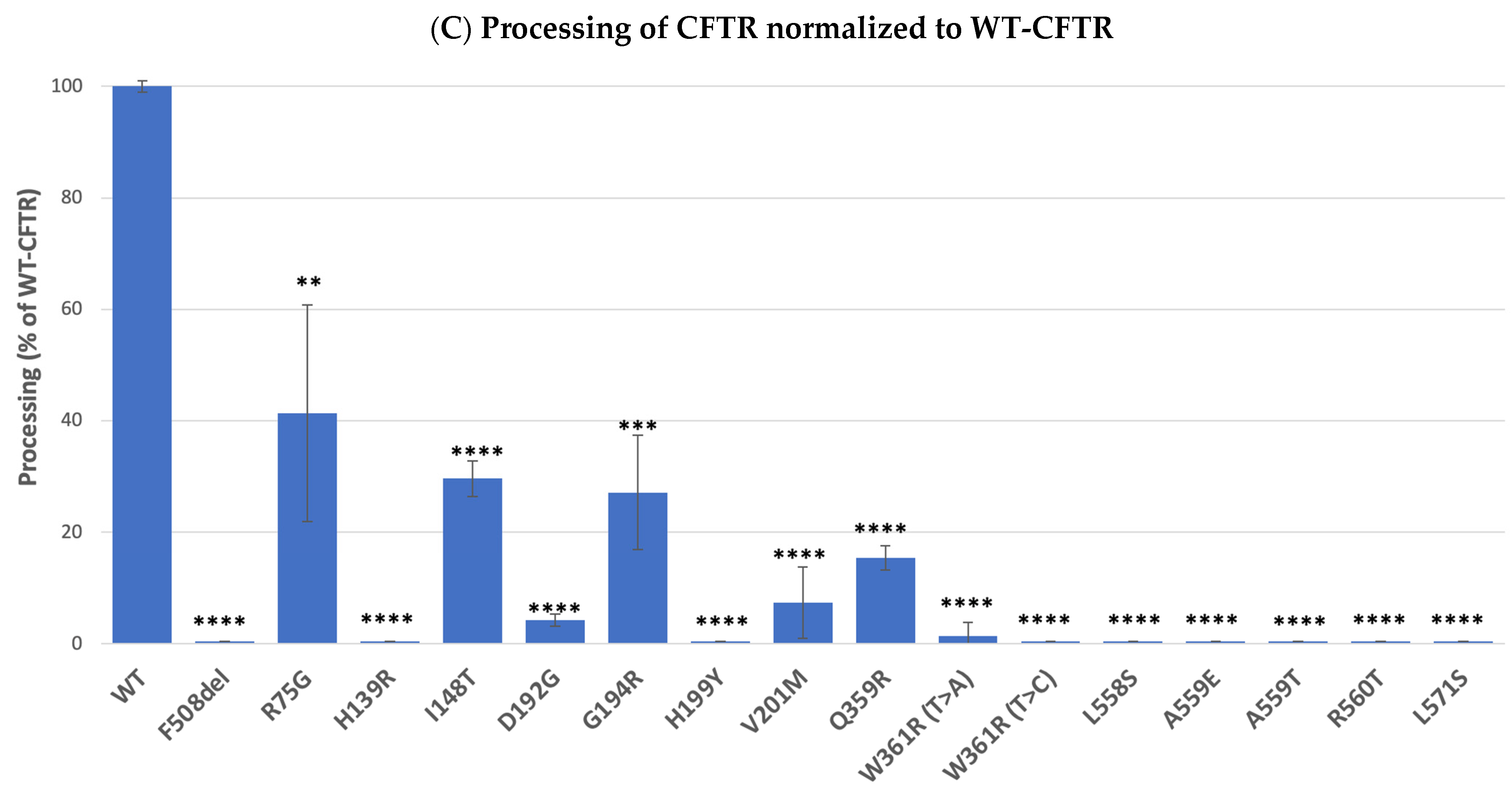
2.2. Impact of Mutations upon CFTR Processing and Response to Correctors
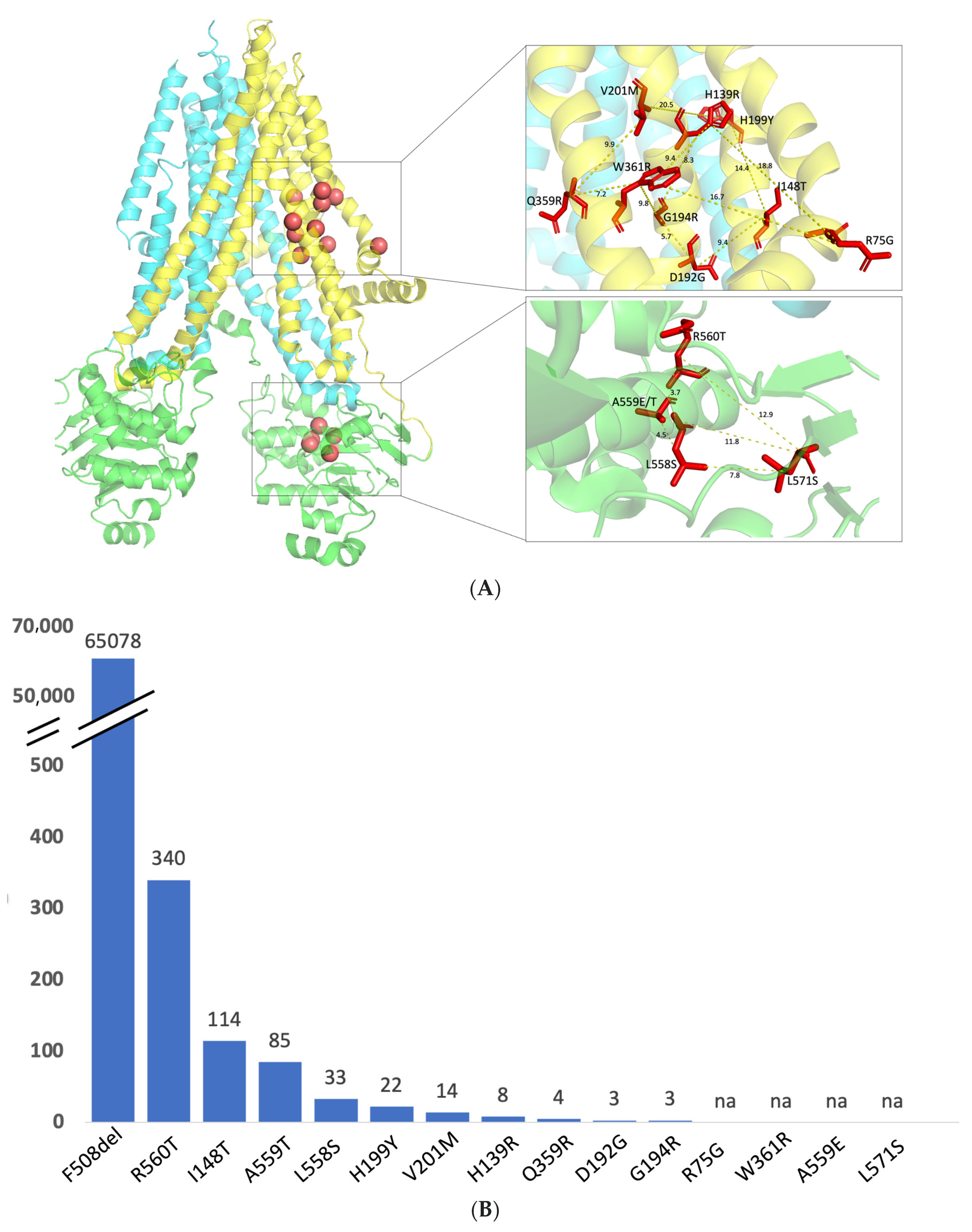
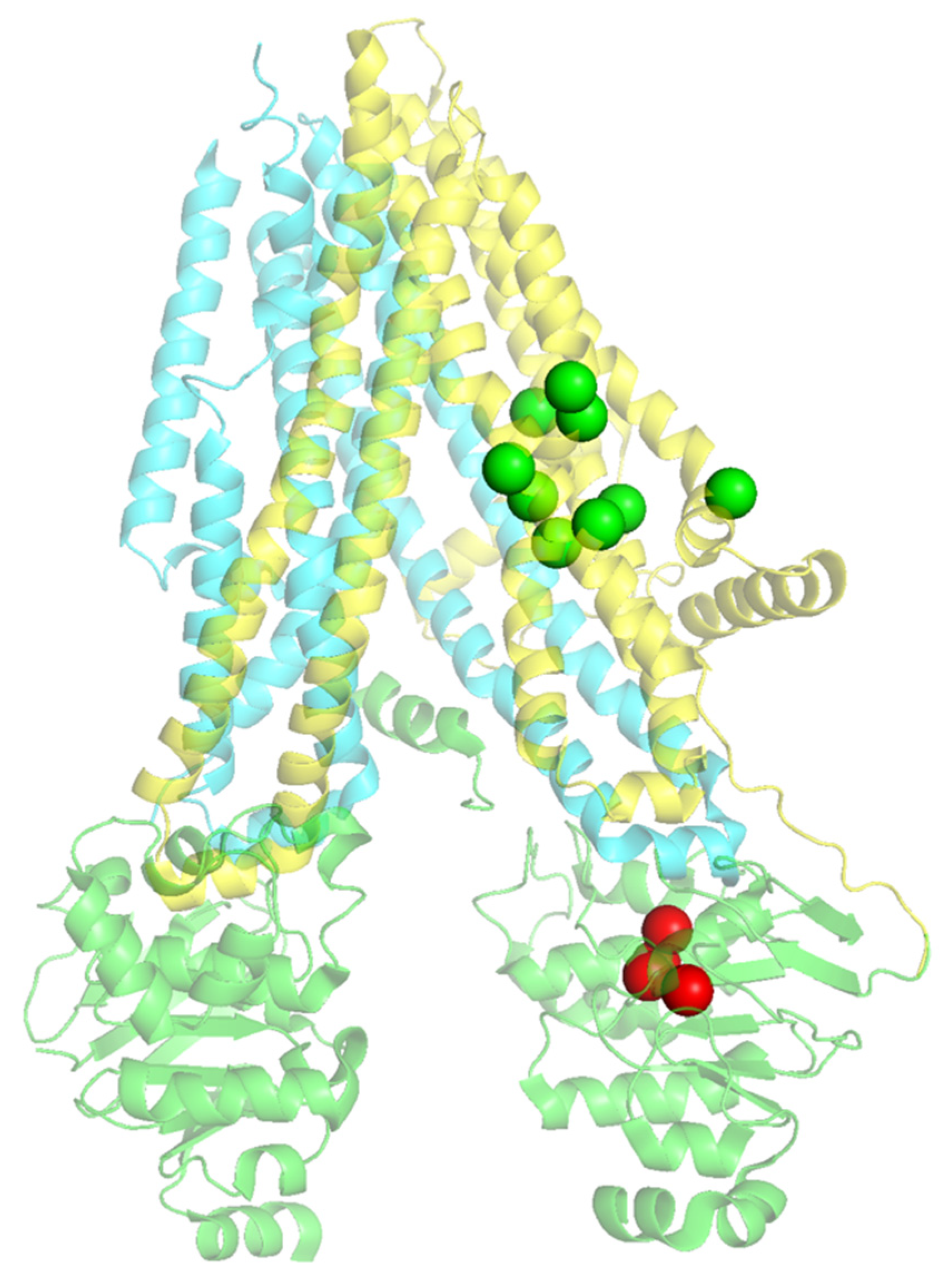
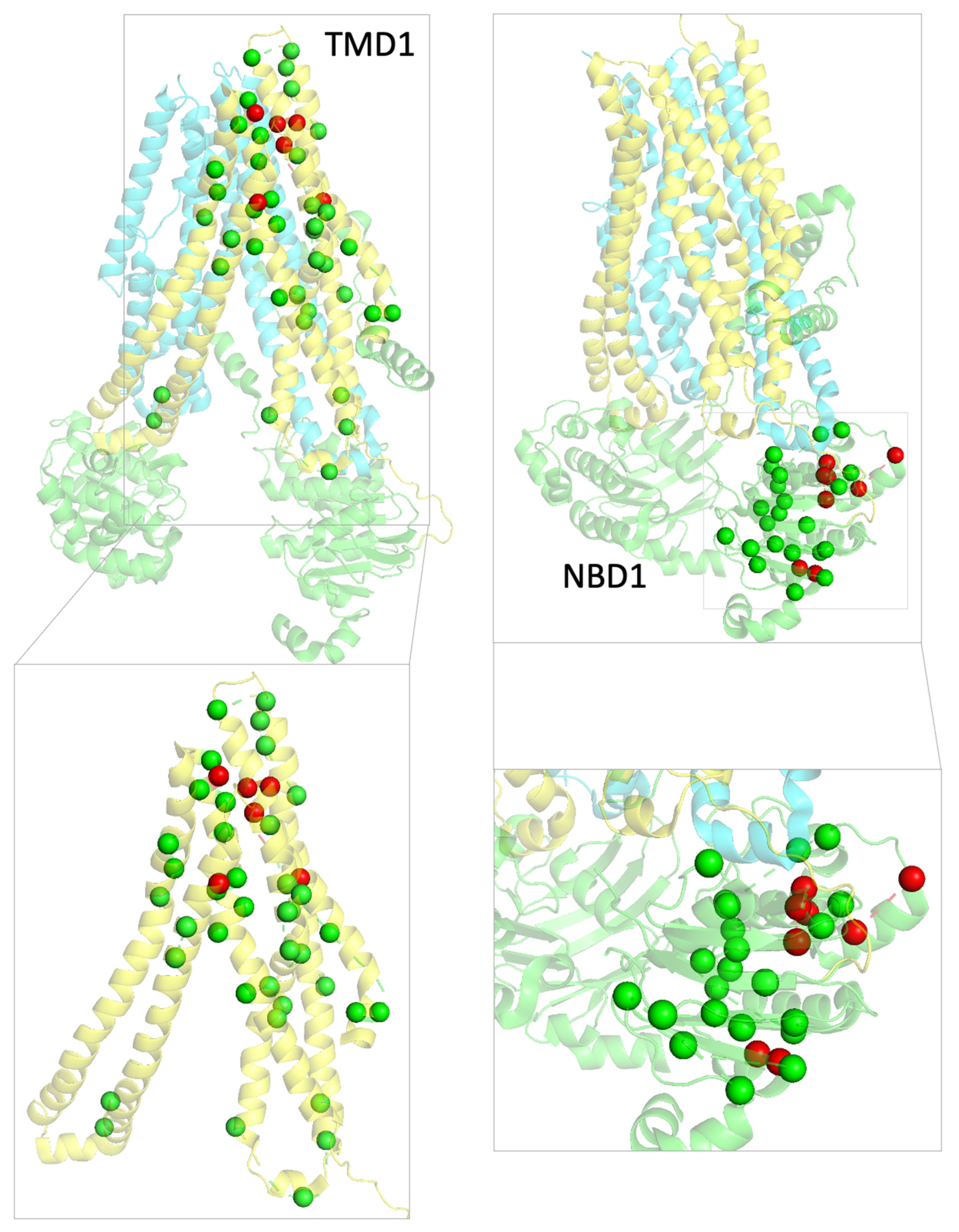
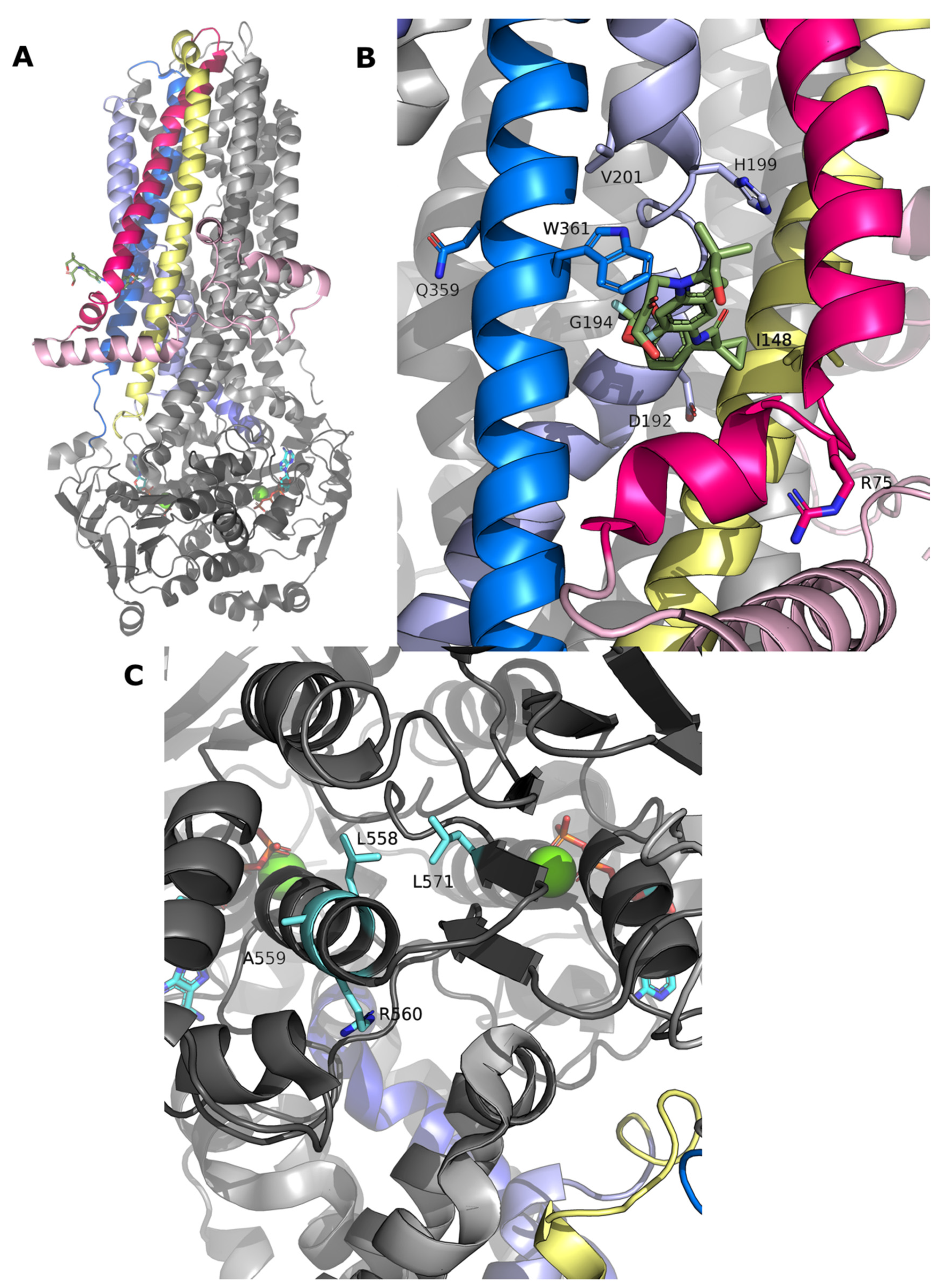
2.3. Structural Mapping of Mutations
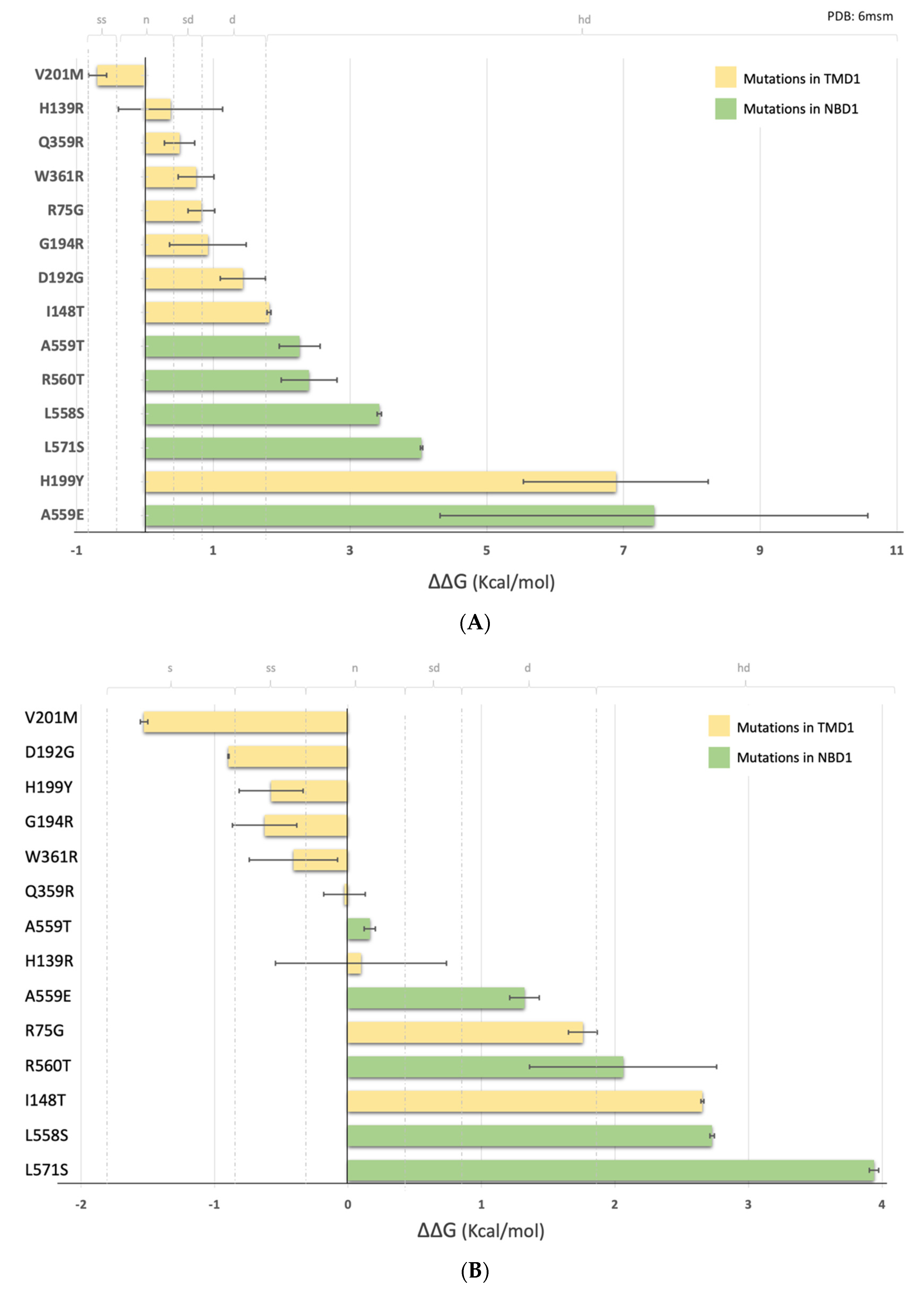
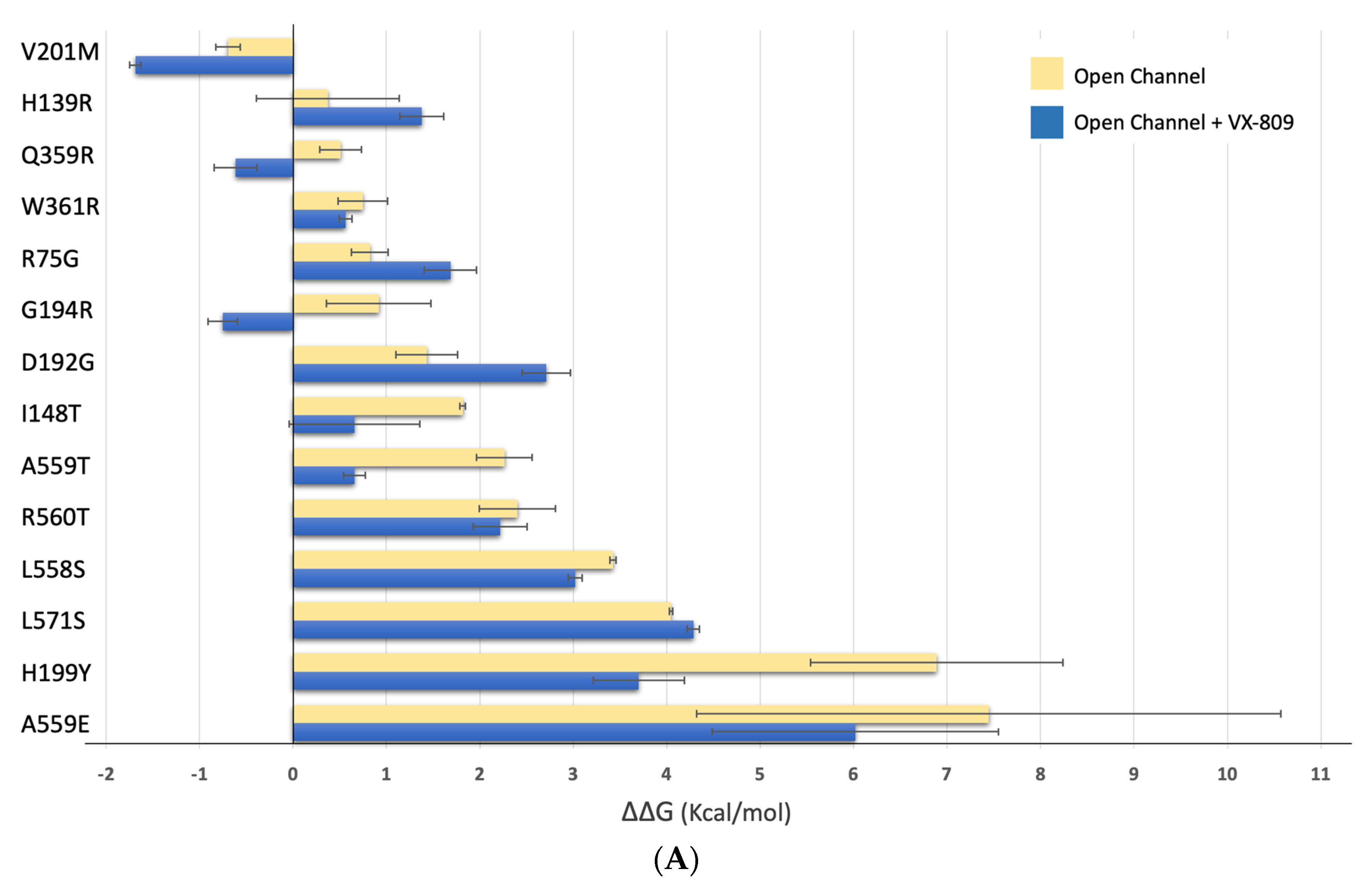
2.4. Impact of the Mutations upon Stability of CFTR Structure
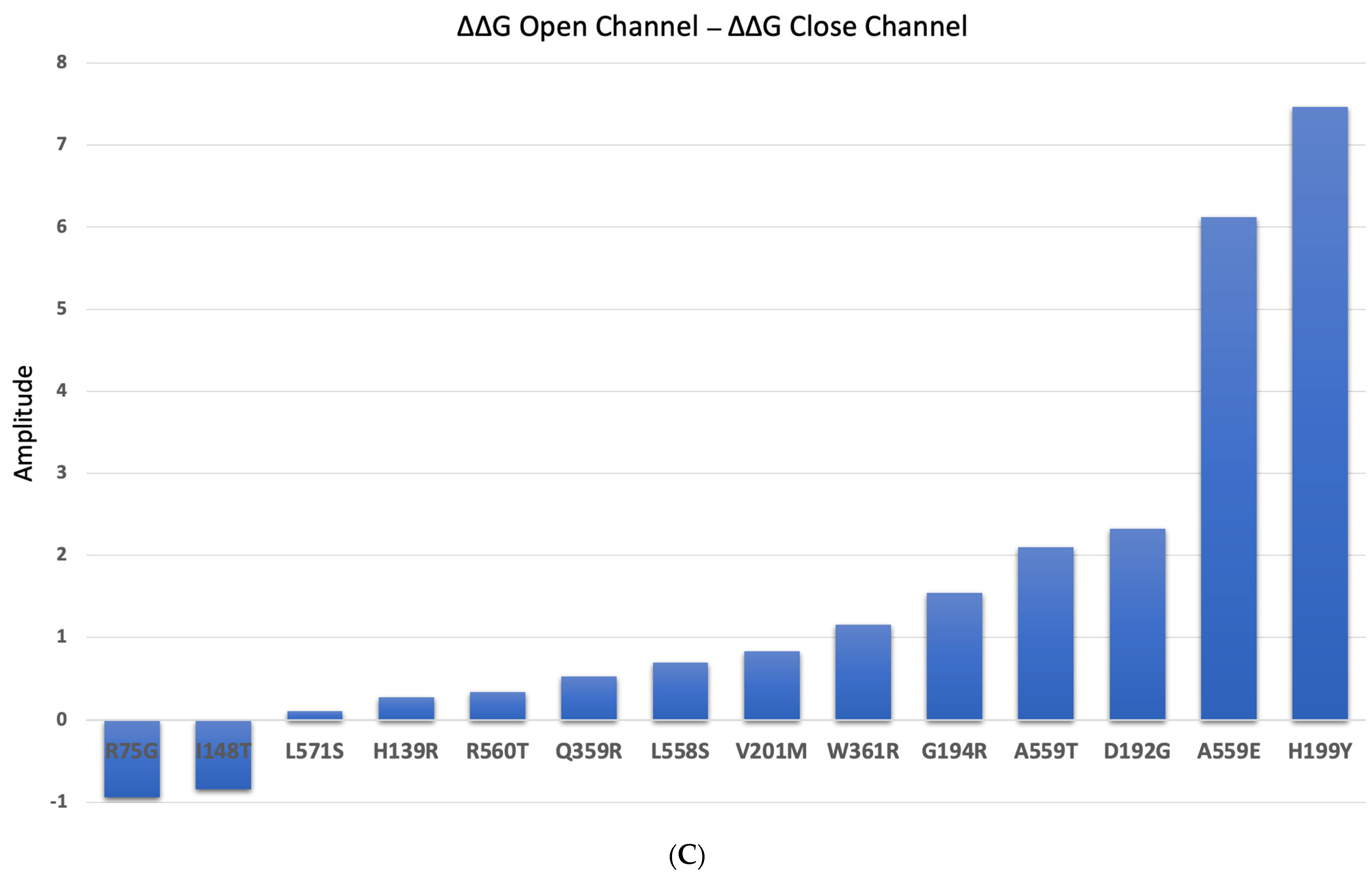
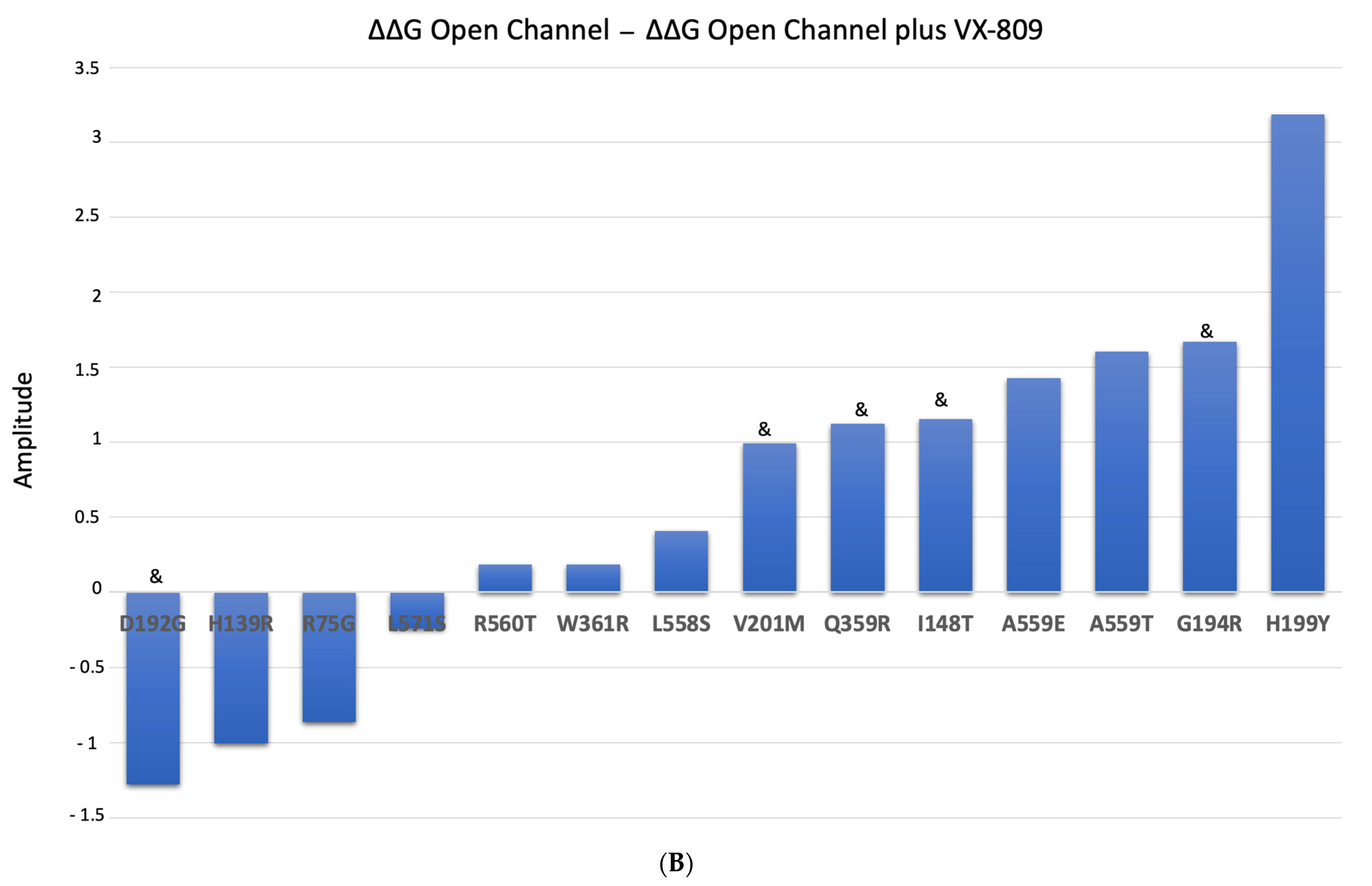
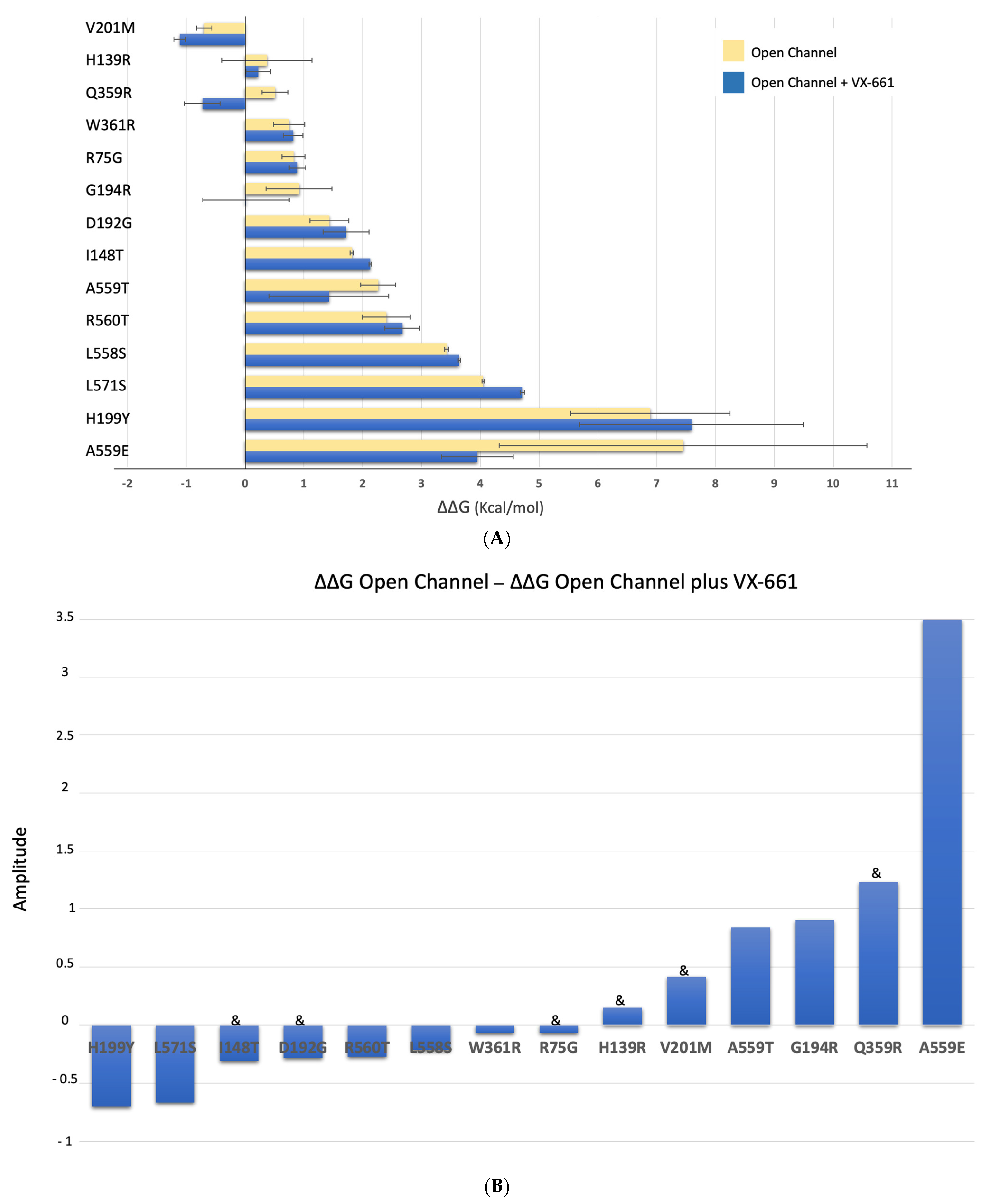
2.5. Impact of the Mutations upon Stability of CFTR Structure in the Presence of Modulators
3. Discussion
4. Methods and Materials
4.1. Site-Directed Mutagenesis
4.2. Cell Lines
4.3. Treatment with CFTR Modulators
4.4. Western Blot Analysis
4.5. Statistical Analyses
4.6. Protein Structure Preparation
4.7. ΔΔG Calculations of CFTR Mutants Using FoldX
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boucher, R.C. Cystic fibrosis: A disease of vulnerability to airway surface dehydration. Trends Mol. Med. 2007, 13, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ferec, C.; Cutting, G.R. Assessing the Disease-Liability of Mutations in CFTR. Cold. Spring Harb. Perspect. Med. 2012, 2, a009480. [Google Scholar] [CrossRef]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Infield, D.T.; Strickland, K.M.; Gaggar, A.; McCarty, N.A. The molecular evolution of function in the CFTR chloride channel. J. Gen. Physiol. 2021, 153, 12625. [Google Scholar] [CrossRef] [PubMed]
- Mornon, J.P.; Lehn, P.; Callebaut, I. Atomic model of human cystic fibrosis transmembrane conductance regulator: Membrane-spanning domains and coupling interfaces. Cell Mol. Life Sci. 2008, 65, 2594–2612. [Google Scholar] [CrossRef]
- Dalton, J.; Kalid, O.; Schushan, M.; Ben-Tal, N.; Villa-Freixa, J. New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J. Chem. Inf. Model. 2012, 52, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Csanady, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2016, 167, 1586–1597. [Google Scholar] [CrossRef]
- Amaral, M.D.; Kunzelmann, K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharm. Sci. 2007, 28, 334–341. [Google Scholar] [CrossRef]
- Martin, C.; Legeai, C.; Regard, L.; Cantrelle, C.; Dorent, R.; Carlier, N.; Kerbaul, F.; Burgel, P.R. Major Decrease in Lung Transplantation for Patients with Cystic Fibrosis in France. Am. J. Respir. Crit. Care. Med. 2022, 205, 584–586. [Google Scholar] [CrossRef]
- Simmonds, N.J. Is it cystic fibrosis? The challenges of diagnosing cystic fibrosis. Paediatr. Respir. Rev. 2019, 31, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Balch, W.E. Hallmarks of therapeutic management of the cystic fibrosis functional landscape. J. Cyst. Fibros. 2015, 14, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Cozens, A.L.; Yezzi, M.J.; Chin, L.; Simon, E.M.; Friend, D.S.; Gruenert, D.C. Chloride ion transport in transformed normal and cystic fibrosis epithelial cells. Adv. Exp. Med. Biol. 1991, 290, 187–194. [Google Scholar]
- Awatade, N.T.; Uliyakina, I.; Farinha, C.M.; Clarke, L.A.; Mendes, K.; Sole, A.; Pastor, J.; Ramos, M.M.; Amaral, M.D. Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis. EBioMedicine 2015, 2, 147–153. [Google Scholar] [CrossRef]
- Han, S.T.; Rab, A.; Pellicore, M.J.; Davis, E.F.; McCague, A.F.; Evans, T.A.; Joynt, A.T.; Lu, Z.; Cai, Z.; Raraigh, K.S.; et al. Residual function of cystic fibrosis mutants predicts response to small molecule CFTR modulators. J. CI Insight 2018, 3, 121159. [Google Scholar] [CrossRef]
- He, L.; Kota, P.; Aleksandrov, A.A.; Cui, L.; Jensen, T.; Dokholyan, N.V.; Riordan, J.R. Correctors of {Delta}F508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013, 27, 536–545. [Google Scholar] [CrossRef]
- Seibert, F.S.; Jia, Y.; Mathews, C.J.; Hanrahan, J.W.; Riordan, J.R.; Loo, T.W.; Clarke, D.M. Disease-associated mutations in cytoplasmic loops 1 and 2 of cystic fibrosis transmembrane conductance regulator impede processing or opening of the channel. Biochemistry 1997, 36, 11966–11974. [Google Scholar] [CrossRef]
- Baatallah, N.; Elbahnsi, A.; Mornon, J.P.; Chevalier, B.; Pranke, I.; Servel, N.; Zelli, R.; Decout, J.L.; Edelman, A.; Sermet-Gaudelus, I.; et al. Pharmacological chaperones improve intra-domain stability and inter-domain assembly via distinct binding sites to rescue misfolded CFTR. Cell Mol. Life Sci. 2021, 78, 7813–7829. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168. [Google Scholar] [CrossRef]
- Billet, A.; Elbahnsi, A.; Jollivet-Souchet, M.; Hoffmann, B.; Mornon, J.P.; Callebaut, I.; Becq, F. Functional and Pharmacological Characterization of the Rare CFTR Mutation W361R. Front. Pharm. 2020, 11, 295. [Google Scholar] [CrossRef]
- Shishido, H.; Yoon, J.S.; Yang, Z.; Skach, W.R. CFTR trafficking mutations disrupt cotranslational protein folding by targeting biosynthetic intermediates. Nat. Commun. 2020, 11, 4258. [Google Scholar] [CrossRef]
- Gregory, R.J.; Rich, D.P.; Cheng, S.H.; Souza, D.W.; Paul, S.; Manavalan, P.; Anderson, M.P.; Welsh, M.J.; Smith, A.E. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol. Cell Biol. 1991, 11, 3886–3893. [Google Scholar] [PubMed]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef]
- Rabeh, W.M.; Bossard, F.; Xu, H.; Okiyoneda, T.; Bagdany, M.; Mulvihill, C.M.; Du, K.; di Bernardo, S.; Liu, Y.; Konermann, L.; et al. Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell 2012, 148, 150–163. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Bahia, M.S.; Khazanov, N.; Zhou, Q.; Yang, Z.; Wang, C.; Hong, J.S.; Rab, A.; Sorscher, E.J.; Brouillette, C.G.; Hunt, J.F.; et al. Stability Prediction for Mutations in the Cytosolic Domains of Cystic Fibrosis Transmembrane Conductance Regulator. J. Chem. Inf. Model. 2021, 61, 1762–1777. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Hoffmann, B.; Lehn, P.; Mornon, J.P. Molecular modelling and molecular dynamics of CFTR. Cell Mol. Life Sci. 2017, 74, 3–22. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Delta508 CFTR by Trikafta modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Rapino, D.; Cebotaru, L. Combination of Correctors Rescues CFTR Transmembrane-Domain Mutants by Mitigating their Interactions with Proteostasis. Cell Physiol. Biochem. 2017, 41, 2194–2210. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Rodrigues, C.H.M.; Ascher, D.B. mCSM-membrane: Predicting the effects of mutations on transmembrane proteins. Nucleic Acids Res. 2020, 48, W147–W153. [Google Scholar] [CrossRef]
- Angles, F.; Wang, C.; Balch, W.E. Spatial covariance analysis reveals the residue-by-residue thermodynamic contribution of variation to the CFTR fold. Commun. Biol. 2022, 5, 356. [Google Scholar] [CrossRef]
- Wang, C.; Angles, F.; Balch, W.E. Triangulating variation in the population to define mechanisms for precision management of genetic disease. Structure 2022, 30, 1190–1207.e5. [Google Scholar] [CrossRef] [PubMed]
- Cozens, A.L.; Yezzi, M.J.; Kunzelmann, K.; Ohrui, T.; Chin, L.; Eng, K.; Finkbeiner, W.E.; Widdicombe, J.H.; Gruenert, D.C. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994, 10, 38–47. [Google Scholar] [CrossRef]
- Awatade, N.T.; Ramalho, S.; Silva, I.A.L.; Felicio, V.; Botelho, H.M.; de Poel, E.; Vonk, A.; Beekman, J.M.; Farinha, C.M.; Amaral, M.D. R560S: A class II CFTR mutation that is not rescued by current modulators. J. Cyst. Fibros. 2019, 18, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, S.S.; Silva, I.A.L.; Amaral, M.D.; Farinha, C.M. Rare Trafficking CFTR Mutations Involve Distinct Cellular Retention Machineries and Require Different Rescuing Strategies. Int. J. Mol. Sci. 2021, 23, 24. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Reis, P.; Vila-Vicosa, D.; Rocchia, W.; Machuqueiro, M. PypKa: A Flexible Python Module for Poisson-Boltzmann-Based pK(a) Calculations. J. Chem. Inf. Model. 2020, 60, 4442–4448. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Ferec, C.; Novelli, G.; Verlingue, C.; Quere, I.; Dallapiccola, B.; Audrezet, M.P.; Mercier, B. Identification of six novel CFTR mutations in a sample of Italian cystic fibrosis patients. Mol. Cell Probes. 1995, 9, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Rescue of DeltaF508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol. Pharm. 2005, 2, 407–413. [Google Scholar] [CrossRef]
- Bozon, D.; Zielenski, J.; Rininsland, F.; Tsui, L.C. Identification of four new mutations in the cystic fibrosis transmembrane conductance regulator gene: I148T, L1077P, Y1092X, 2183AA- ->G. Hum.Mutat. 1994, 3, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, G.; Rippa, E.; Raia, V.; Salvatore, D.; Massa, C.; de Ritis, G.; Salvatore, F. Clinical features of cystic fibrosis patients with rare genotypes. J. Med. Genet. 1996, 33, 73–76. [Google Scholar] [CrossRef]
- Population variation of common cystic fibrosis mutations. The Cystic Fibrosis Genetic Analysis Consortium. Hum. Mutat. 1994, 4, 167–177. [CrossRef]
- Rohlfs, E.M.; Zhou, Z.; Sugarman, E.A.; Heim, R.A.; Pace, R.G.; Knowles, M.R.; Silverman, L.M.; Allitto, B.A. The I148T CFTR allele occurs on multiple haplotypes: A complex allele is associated with cystic fibrosis. Genet. Med. 2002, 4, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, K.G.; Highsmith, W.E.; Amos, J.; Pratt, V.M.; Roa, B.; Friez, M.; Pike-Buchanan, L.L.; Buyse, I.M.; Redman, J.B.; Strom, C.M.; et al. Genotype-phenotype correlation and frequency of the 3199del6 cystic fibrosis mutation among I148T carriers: Results from a collaborative study. Genet. Med. 2004, 6, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, D.; Pepe, A.; Carnovale, V.; Majo, F.; Padoan, R.; Quattrucci, S.; Salvatore, M.; Taruscio, D.; Amato, A.; Ferrari, G.; et al. Elexacaftor/tezacaftor/ivacaftor for CFTR variants giving rise to diagnostic uncertainty: Personalised medicine or over-medicalisation? J. Cyst. Fibros 2022, 21, 544–548. [Google Scholar] [CrossRef]
- Caputo, A.; Hinzpeter, A.; Caci, E.; Pedemonte, N.; Arous, N.; Di Duca, M.; Zegarra-Moran, O.; Fanen, P.; Galietta, L.J. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J. Pharmacol. Exp. Ther. 2009, 330, 783–791. [Google Scholar] [CrossRef]
- Terlizzi, V.; Castaldo, G.; Salvatore, D.; Lucarelli, M.; Raia, V.; Angioni, A.; Carnovale, V.; Cirilli, N.; Casciaro, R.; Colombo, C.; et al. Genotype-phenotype correlation and functional studies in patients with cystic fibrosis bearing CFTR complex alleles. J. Med. Genet. 2017, 54, 224–235. [Google Scholar] [CrossRef]
- Choi, J.Y.; Muallem, D.; Kiselyov, K.; Lee, M.G.; Thomas, P.J.; Muallem, S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 2001, 410, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Audrezet, M.P.; Canki-Klain, N.; Mercier, B.; Bracar, D.; Verlingue, C.; Ferec, C. Identification of three novel mutations (457 TAT-->G, D192G, Q685X) in the Slovenian CF patients. Hum. Genet. 1994, 93, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Zybert, K.; Wozniacki, L.; Tomaszewska-Sobczynska, A.; Wertheim-Tysarowska, K.; Czerska, K.; Oltarzewski, M.; Sands, D. Clinical characteristics of rare CFTR mutations causing cystic fibrosis in Polish population. Pediatr. Pulmonol. 2020, 55, 2097–2107. [Google Scholar] [CrossRef]
- Keiles, S.; Kammesheidt, A. Identification of CFTR, PRSS1, and SPINK1 mutations in 381 patients with pancreatitis. Pancreas 2006, 33, 221–227. [Google Scholar] [CrossRef]
- Decaestecker, K.; Decaestecker, E.; Castellani, C.; Jaspers, M.; Cuppens, H.; De Boeck, K. Genotype/phenotype correlation of the G85E mutation in a large cohort of cystic fibrosis patients. Eur. Respir. J. 2004, 23, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, A.L.; Ferri, A.; Passos-Bueno, M.R.; Kim, C.E.; Nakaie, C.M.; Gomes, C.E.; Damaceno, N.; Zatz, M. Molecular analysis in Brazilian cystic fibrosis patients reveals five novel mutations. Genet. Test. 2000, 4, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Alper, O.M.; Lu, J.F.; Wang, S.P.; Guo, L.; Chiang, H.S.; Wong, L.J. Mutation spectrum of the CFTR gene in Taiwanese patients with congenital bilateral absence of the vas deferens. Hum. Reprod. 2005, 20, 2470–2475. [Google Scholar] [CrossRef]
- Danziger, K.L.; Black, L.D.; Keiles, S.B.; Kammesheidt, A.; Turek, P.J. Improved detection of cystic fibrosis mutations in infertility patients with DNA sequence analysis. Hum. Reprod. 2004, 19, 540–546. [Google Scholar] [CrossRef]
- Boudaya, M.; Fredj, S.H.; Haj, R.B.; Khrouf, M.; Bouker, A.; Halouani, L.; Messaoud, T. Cystic fibrosis transmembrane conductance regulator mutations and polymorphisms associated with congenital bilateral absence of vas deferens in a restricted group of patients from North Africa. Ann. Hum. Biol. 2012, 39, 76–79. [Google Scholar] [CrossRef]
- Raraigh, K.S.; Lewis, M.H.; Collaco, J.M.; Corey, M.; Penland, C.M.; Stephenson, A.L.; Rommens, J.M.; Castellani, C.; Cutting, G.R. Caution advised in the use of CFTR modulator treatment for individuals harboring specific CFTR variants. J. Cyst. Fibros 2022, 21, 856–860. [Google Scholar] [CrossRef]
- Arora, K.; Yang, F.; Brewington, J.; McPhail, G.; Cortez, A.R.; Sundaram, N.; Ramananda, Y.; Ogden, H.; Helmrath, M.; Clancy, J.P.; et al. Patient personalized translational tools in cystic fibrosis to transform data from bench to bed-side and back. Am. J. Physiol. Gastrointest Liver Physiol. 2021, 320, G1123–G1130. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, T.; Hubert, D.; Fonknechten, N.; Dusser, D.; Kaplan, J.C.; Beldjord, C. Unexpected inactivation of acceptor consensus splice sequence by a -3 C to T transition in intron 2 of the CFTR gene. Hum. Genet. 1994, 94, 65–68. [Google Scholar] [CrossRef]
- Alonso, M.J.; Heine-Suner, D.; Calvo, M.; Rosell, J.; Gimenez, J.; Ramos, M.D.; Telleria, J.J.; Palacio, A.; Estivill, X.; Casals, T. Spectrum of mutations in the CFTR gene in cystic fibrosis patients of Spanish ancestry. Ann. Hum. Genet. 2007, 71, 194–201. [Google Scholar] [CrossRef]
- Kanavakis, E.; Tzetis, M.; Antoniadi, T.; Traeger-Synodinos, J.; Doudounakis, S.; Adam, G.; Matsaniotis, N.; Kattamis, C. Mutation analysis of ten exons of the CFTR gene in Greek cystic fibrosis patients: Characterization of 74.5% of CF alleles including one novel mutation. Hum. Genet. 1995, 96, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Strandvik, B.; Bjorck, E.; Fallstrom, M.; Gronowitz, E.; Thountzouris, J.; Lindblad, A.; Markiewicz, D.; Wahlstrom, J.; Tsui, L.C.; Zielenski, J. Spectrum of mutations in the CFTR gene of patients with classical and atypical forms of cystic fibrosis from southwestern Sweden: Identification of 12 novel mutations. Genet. Test. 2001, 5, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Pepper, M.S. Cystic Fibrosis in the African Diaspora. Ann. Am. Thorac. Soc. 2017, 14, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vouk, K.; Strmecki, L.; Liovic, M.; Kopriva, S.; Micetic-Turk, D.; Komel, R. Mutational analysis of 30 Slovenian cystic fibrosis patients compared to known Slovenian and European CF mutation spectra. Pflug. Arch. 2000, 439, R63–R65. [Google Scholar] [CrossRef]
- Castellani, C.; Cuppens, H.; Macek, M., Jr.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros 2008, 7, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Roxo-Rosa, M.; Xu, Z.; Schmidt, A.; Neto, M.; Cai, Z.; Soares, C.M.; Sheppard, D.N.; Amaral, M.D. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 17891–17896. [Google Scholar] [CrossRef]
- Lewis, H.A.; Zhao, X.; Wang, C.; Sauder, J.M.; Rooney, I.; Noland, B.W.; Lorimer, D.; Kearins, M.C.; Conners, K.; Condon, B.; et al. Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J. Biol. Chem. 2005, 280, 1346–1353. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Sabirzhanova, I.; Rapino, D.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Correctors Rescue CFTR Mutations in Nucleotide-Binding Domain 1 (NBD1) by Modulating Proteostasis. Chembiochem 2016, 17, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLOSED | “OPEN” | OPEN + VX-809 | OPEN + VX-661 | ||
|---|---|---|---|---|---|
| PDB | 5uak | 6msm | 7svd | 7sv7 | |
| Mutation | |||||
| TMD1 | R75G | 1.760 ± 0.108 | 0.824 ± 0.196 | 1.685 ± 0.281 | 0.890 ± 0.140 |
| H139R | 0.097 ± 0.641 | 0.374 ± 0.763 | 1.379 ± 0.236 | 0.224 ± 0.212 | |
| I148T | 2.654 ± 0.010 | 1.817 ± 0.029 | 0.661 ± 0.699 | 2.127 ± 0.020 | |
| D192G | −0.895 ± 0.003 | 1.433 ± 0.331 | 2.711 ± 0.258 | 1.716 ± 0.390 | |
| G194R | −0.624 ± 0.241 | 0.919 ± 0.560 | −0.751 ± 0.155 | 0.015 ± 0.735 | |
| H199Y | −0.576 ± 0.240 | 6.888 ± 1.351 | 3.700 ± 0.489 | 7.591 ± 1.902 | |
| V201M | −1.527 ± 0.028 | −0.693 ± 0.131 | −1.687 ± 0.061 | −1.109 ± 0.099 | |
| Q359R | −0.026 ± 0.156 | 0.508 ± 0.223 | −0.616 ± 0.230 | −0.724 ± 0.303 | |
| W361R | −0.408 ± 0.331 | 0.749 ± 0.263 | 0.564 ± 0.067 | 0.817 ± 0.168 | |
| NBD1 | L558S | 2.726 ± 0.017 | 3.426 ± 0.032 | 3.020 ± 0.073 | 3.641 ± 0.017 |
| A559T | 0.163 ± 0.041 | 2.263 ± 0.299 | 0.659 ± 0.118 | 1.425 ± 1.014 | |
| A559E | 1.321 ± 0.111 | 7.446 ± 3.128 | 6.019 ± 1.533 | 3.949 ± 0.611 | |
| R560T | 2.060 ± 0.701 | 2.401 ± 0.408 | 2.216 ± 0.288 | 2.673 ± 0.297 | |
| L571S | 3.939 ± 0.036 | 4.045 ± 0.018 | 4.285 ± 0.067 | 4.712 ± 0.034 | |
| F508del | 9.699 ± 0.012 | 13.294 ± 0.576 | 4.401 ± 0.010 | 15.937 ± 0.119 |
| Mutation | Sequence (5′-3′) | |
|---|---|---|
| R75G | For | CATTAATGCCCTTCGGGGATGTTTTTTCTGGAG |
| Rev | CTCCAGAAAAAACATCCCCGAAGGGCATTAATG | |
| H139R | For | ACACTGCTCCTACGCCCAGCCATTTTTG |
| Rev | CAAAAATGGCTGGGCGTAGGAGCAGTGT | |
| I148T | For | TTTGGCCTTCATCACACTGGAATGCAGATGAGA |
| Rev | TCTCATCTGCATTCCAGTGTGATGAAGGCCAAA | |
| D192G | For | AACCTGAACAAATTTGGTGAAGGACTTGCA |
| Rev | TGCAAGTCCTTCACCAAATTTGTTCAGGTT | |
| G194R | For | CAAATTTGATGAAAGACTTGCATTGGC |
| Rev | GCCAATGCAAGTCTTTCATCAAATTT | |
| H199Y | For | GACTTGCATTGGCATATTTCGTGTGGATCG |
| Rev | CGATCCACACGAAATATGCCAATGCAAGTC | |
| V201M | For | TTGGCACATTTCATGTGGATCGCTCCTTTG |
| Rev | CAAAGGAGCGATCCACATGAAATGTGCCAA | |
| Q359R | For | CCCTGGGCTGTACGAACATGGTATGACTCTCTT |
| Rev | AAGAGAGTCATACCATGTTCGTACAGCCCAGGG | |
| W361R (T- > A) | For | GGGCTGTACAAACAAGGTATGACTCTCTTG |
| Rev | CAAGAGAGTCATACCTTGTTTGTACAGCCC | |
| W361R (T- > C) | For | GGGCTGTACAAACACGGTATGACTCTCTTG |
| Rev | CAAGAGAGTCATACCGTGTTTGTACAGCCC | |
| L558S | For | CGAGCAAGAATTTCTTCAGCAAGAGCAGTATAC |
| Rev | GTATACTGCTCTTGCTGAAGAAATTCTTGCTCG | |
| A559T | For | CGAGCAAGAATTTCTTTAACAAGAGCAGTATACAAAG |
| Rev | CTTTGTATACTGCTCTTGTTAAAGAAATTCTTGCTCG | |
| A559E | For | CGAGCAAGAATTTCTTTAGAAAGAGCAGTATAC |
| Rev | GTATACTGCTCTTTCTAAAGAAATTCTTGCTCG | |
| R560T | For | GCAAGAATTTCTTTAGCAACAGCAGTATACAAAGATGCTG |
| Rev | CAGCATCTTTGTATACTGCTGTTGCTAAAGAAATTCTTGC | |
| L571S | For | GATGCTGATTTGTATTTATCAGACTCTCCTTTTGGATACC |
| Rev | GGTATCCAAAAGGAGAGTCTGATAAATACAAATCAGCATC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zacarias, S.; Batista, M.S.P.; Ramalho, S.S.; Victor, B.L.; Farinha, C.M. Rescue of Rare CFTR Trafficking Mutants Highlights a Structural Location-Dependent Pattern for Correction. Int. J. Mol. Sci. 2023, 24, 3211. https://doi.org/10.3390/ijms24043211
Zacarias S, Batista MSP, Ramalho SS, Victor BL, Farinha CM. Rescue of Rare CFTR Trafficking Mutants Highlights a Structural Location-Dependent Pattern for Correction. International Journal of Molecular Sciences. 2023; 24(4):3211. https://doi.org/10.3390/ijms24043211
Chicago/Turabian StyleZacarias, Sónia, Marta S. P. Batista, Sofia S. Ramalho, Bruno L. Victor, and Carlos M. Farinha. 2023. "Rescue of Rare CFTR Trafficking Mutants Highlights a Structural Location-Dependent Pattern for Correction" International Journal of Molecular Sciences 24, no. 4: 3211. https://doi.org/10.3390/ijms24043211