
Identification of Anoikis-Related Subgroups and Prognosis Model in Liver Hepatocellular Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
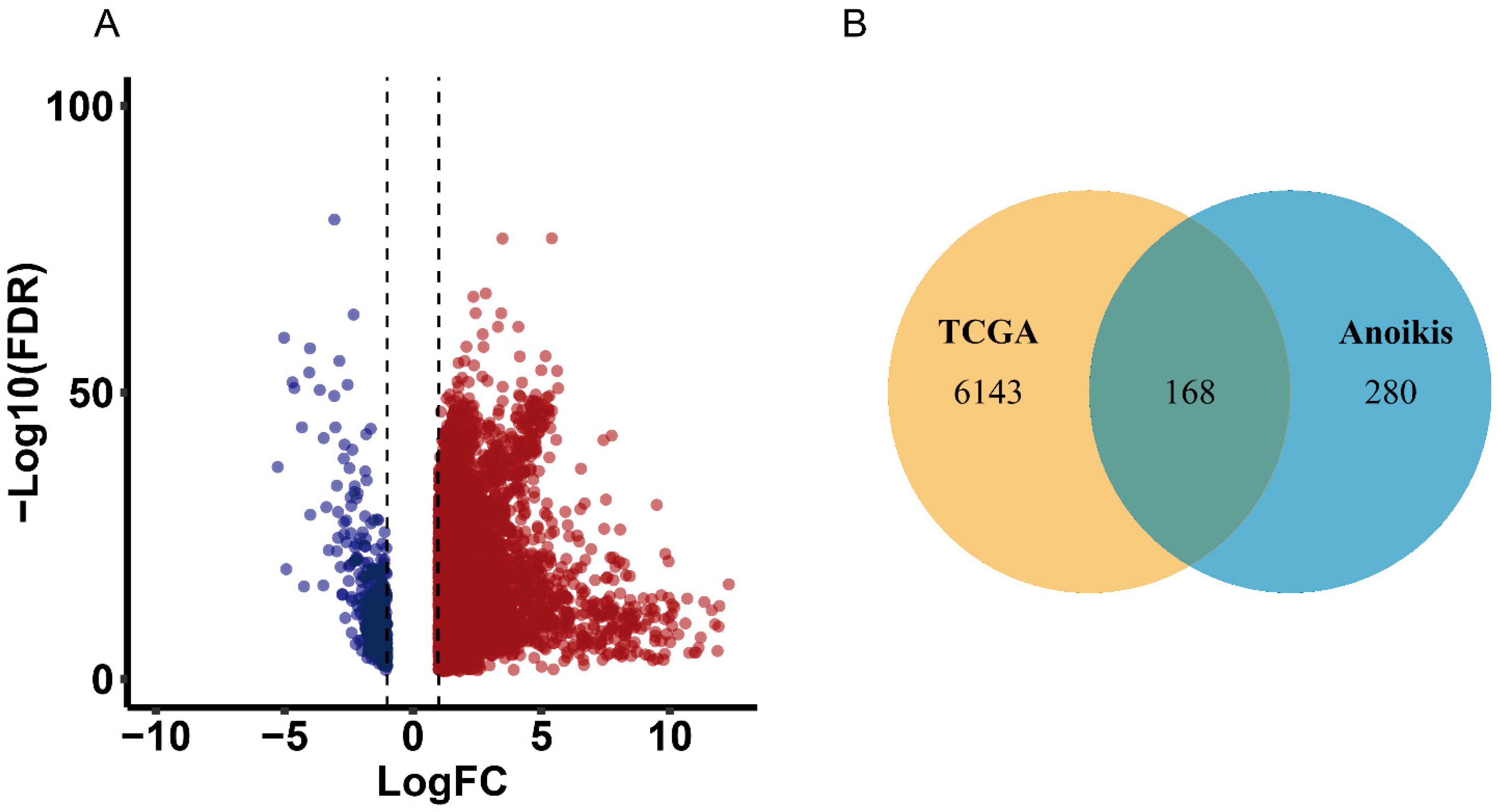
2.1. Identification of Differentially Expressed Overlapping Anoikis-Related Genes
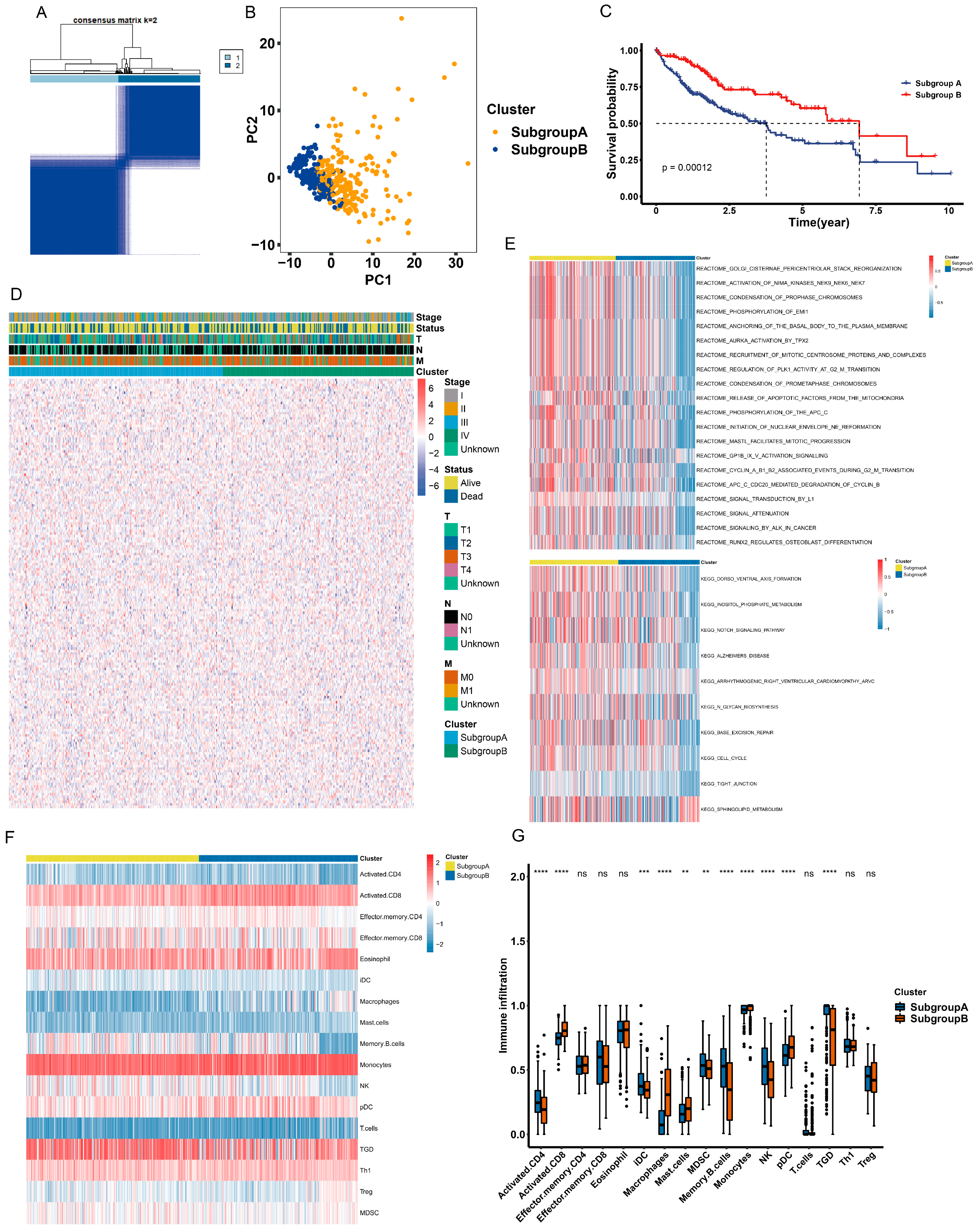
2.2. Identification of Anoikis-Related Subgroups
2.3. Different Characteristics of Biological Behavior between Two Subgroups
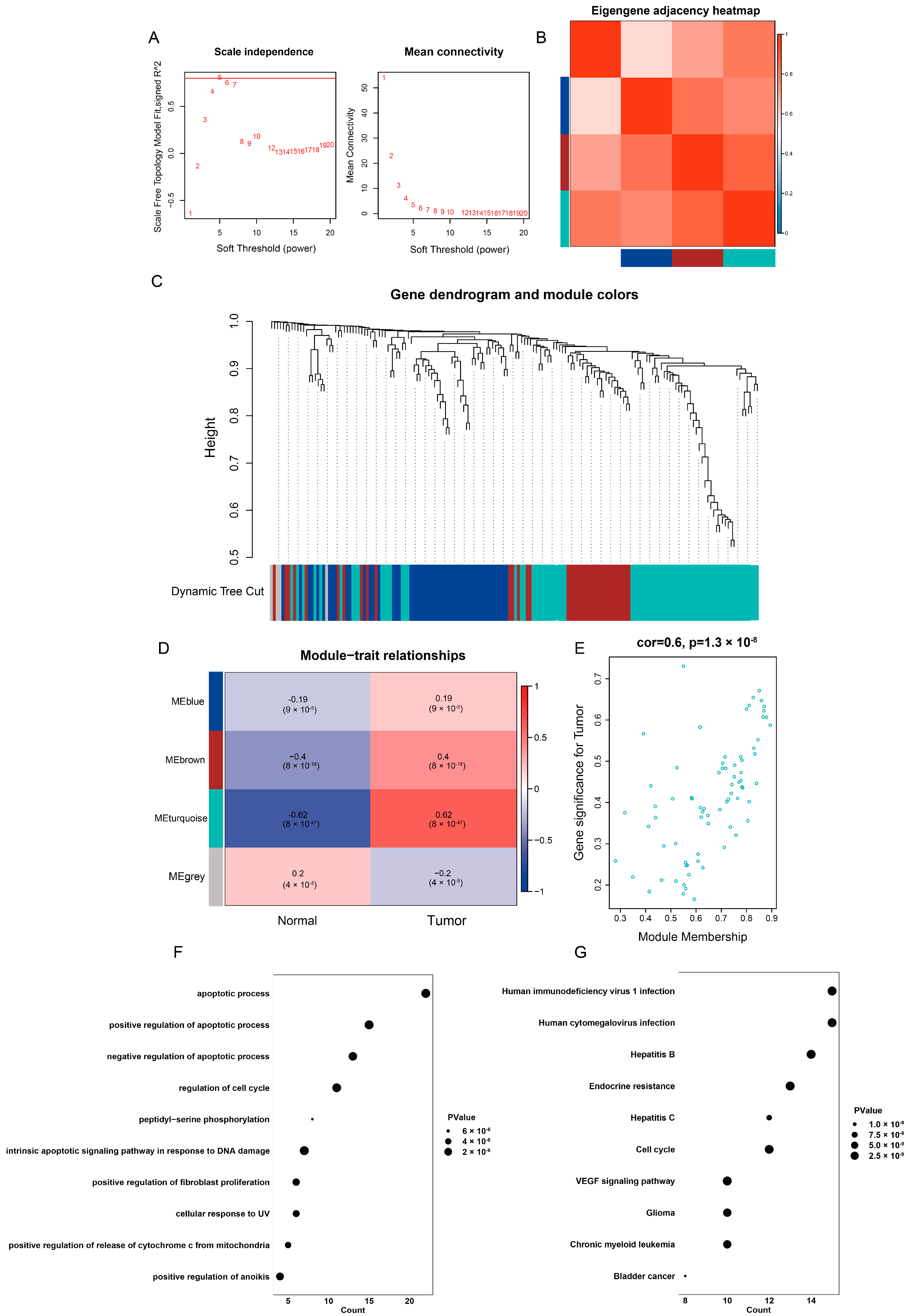
2.4. Identification of Highly Correlated Gene Module in TCGA-LIHC
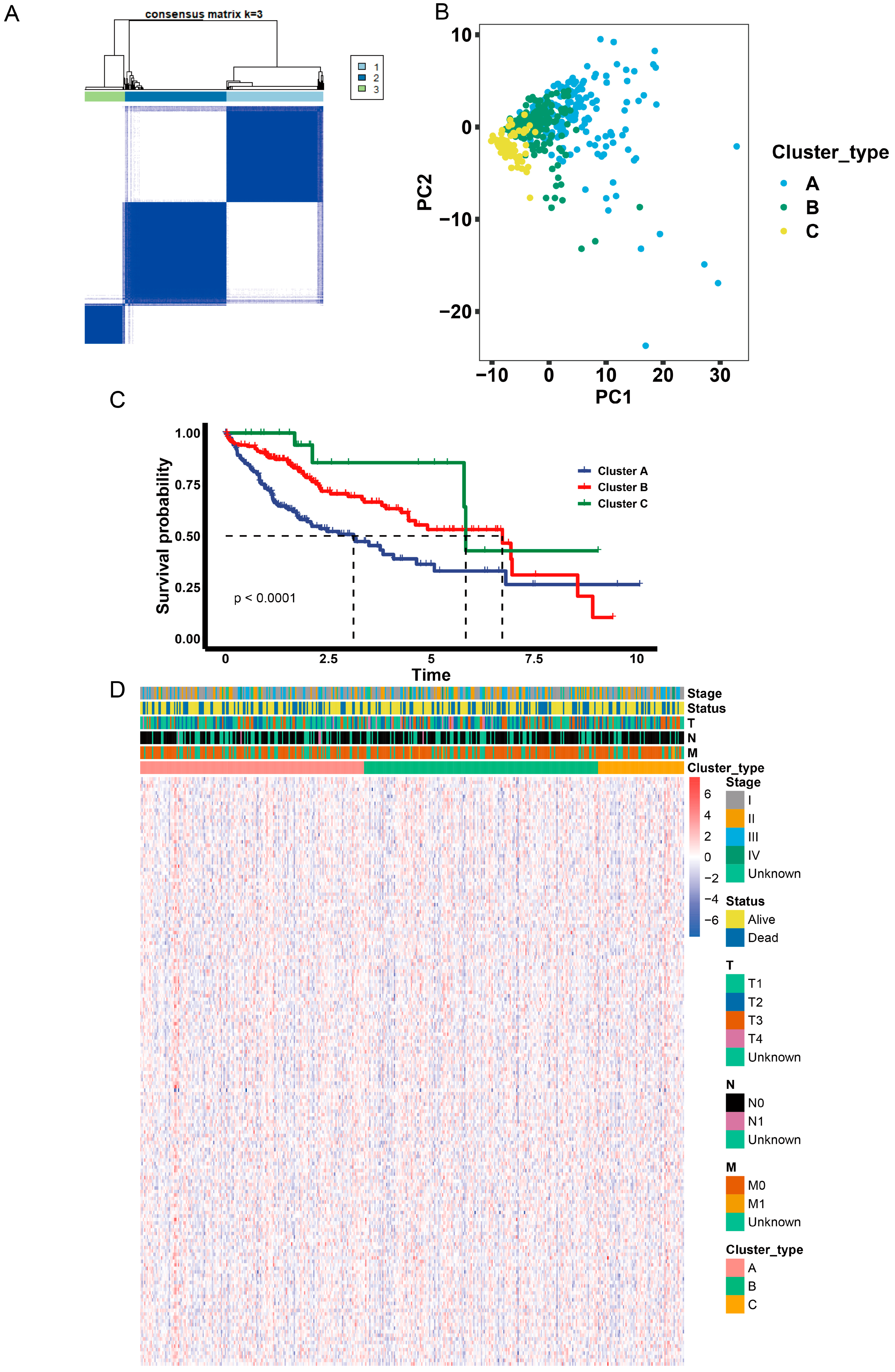
2.5. Identification of Anoikis-Related Gene Clusters
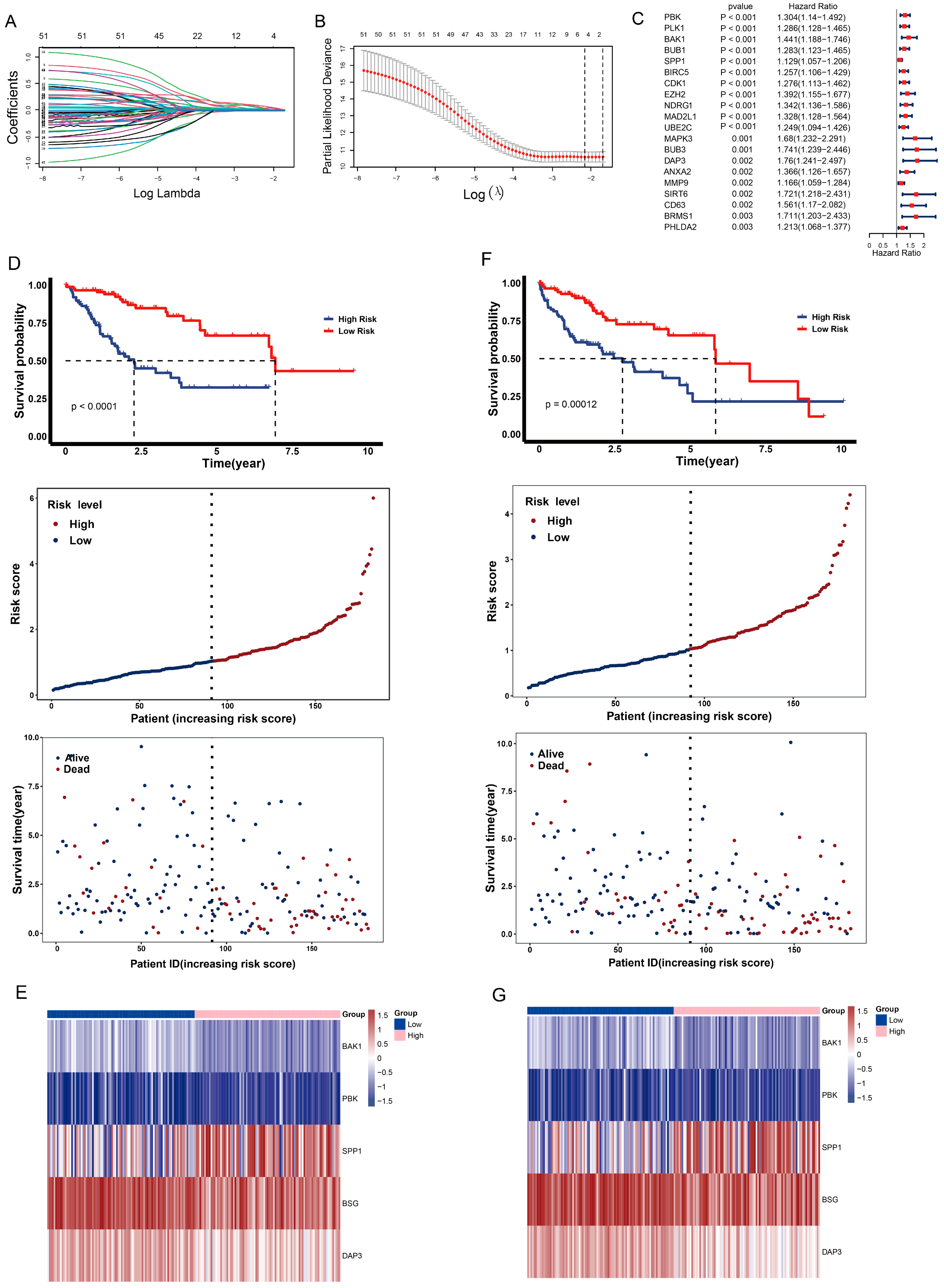
2.6. Construction of Anoikis-Related Prognostic Risk Model
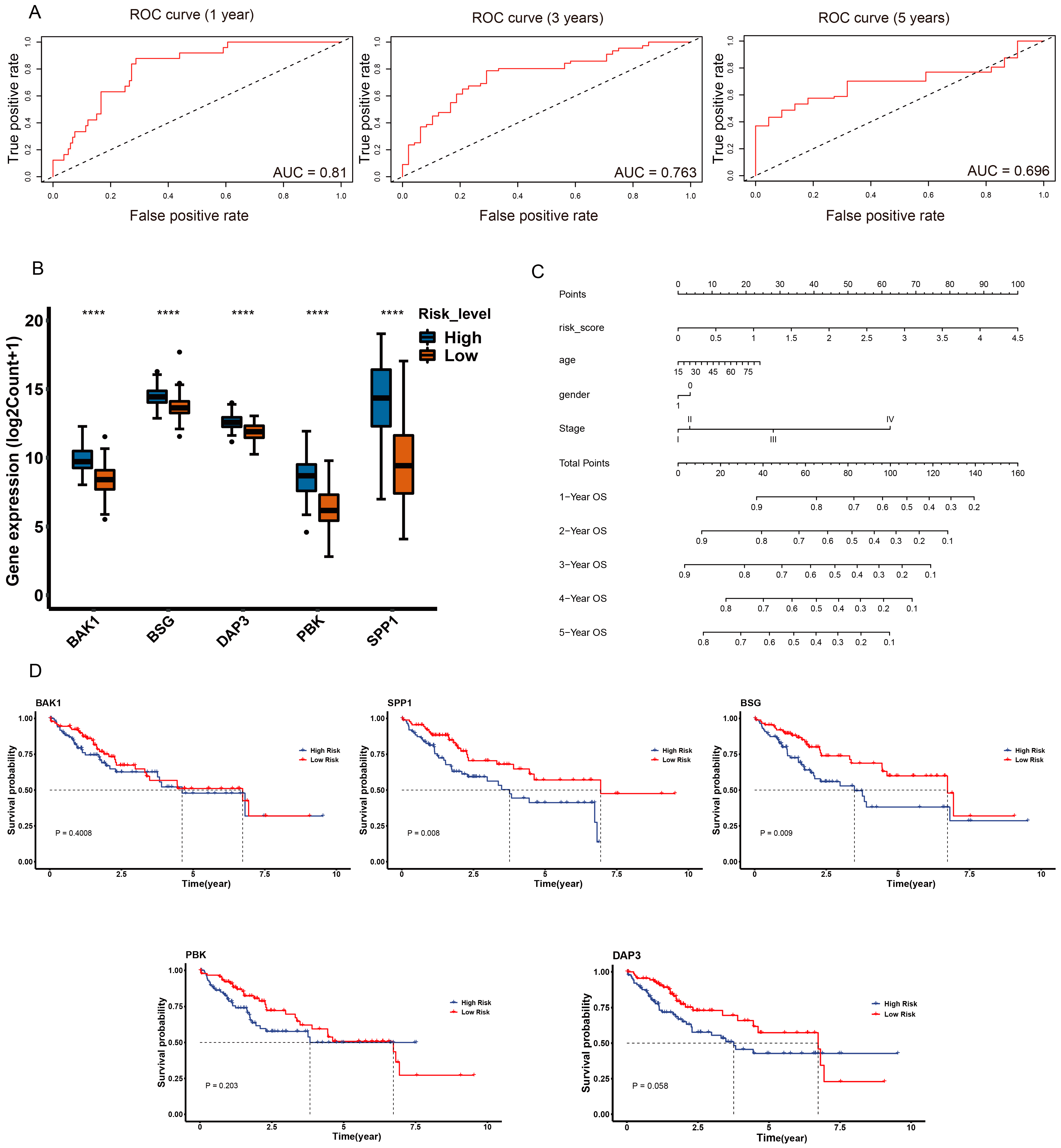
2.7. Validation of Anoikis-Related Prognostic Risk Model
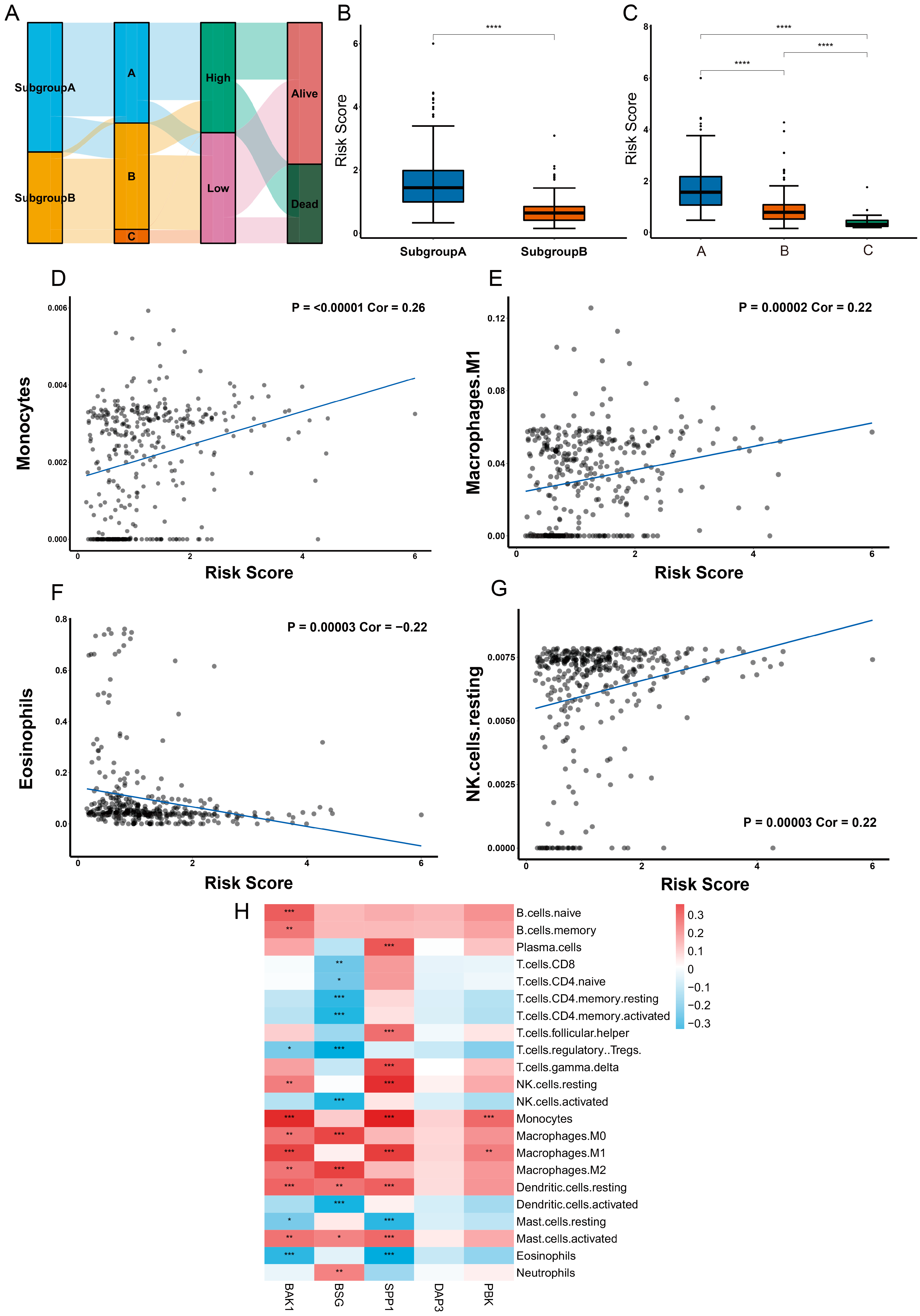
2.8. Different Characteristics of Immune Cell Infiltration between Two Risk Groups
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Weighted Gene Coexpression Network Analysis (WGCNA)
4.3. Consensus Clustering Analysis of Anoikis-Related Genes
4.4. GSVA of Anoikis-Related Genes
4.5. Functional Enrichment Analysis
4.6. Construction of Anoikis-Related Prognostic Risk Model
4.7. Survival Analysis
4.8. Tumor Immune Analysis
4.9. Validation of Prognostic Risk Model
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, D.Q.; Mathurin, P.; Cortez-Pinto, H.; Loomba, R. Global epidemiology of alcohol-associated cirrhosis and HCC: Trends, projections and risk factors. Nat. Rev. Gastroenterol. Hepatol. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cisneros-Garza, L.E.; González-Huezo, M.S.; Moctezuma-Velázquez, C.; Ladrón de Guevara-Cetina, L.; Vilatobá, M.; García-Juárez, I.; Alvarado-Reyes, R.; Álvarez-Treviño, G.A.; Allende-Pérez, S.; Bornstein-Quevedo, L.; et al. The second Mexican consensus on hepatocellular carcinoma. Part I: Epidemiology and diagnosis. Rev. Gastroenterol. Mex. Engl. Ed. 2022, 87, 216–234. [Google Scholar] [CrossRef]
- Yang, J.D.; Heimbach, J.K. New advances in the diagnosis and management of hepatocellular carcinoma. BMJ 2020, 371, m3544. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Zhou, Q.; Dao, D.Y.; Lo, Y.M.D. Circulating biomarkers in the diagnosis and management of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 670–681. [Google Scholar] [CrossRef]
- Raja, A.; Haq, F. Molecular classification of hepatocellular carcinoma: Prognostic importance and clinical applications. J. Cancer Res. Clin. Oncol. 2022, 148, 15–29. [Google Scholar] [CrossRef]
- Adeshakin, F.O.; Adeshakin, A.O.; Afolabi, L.O.; Yan, D.; Zhang, G.; Wan, X. Mechanisms for Modulating Anoikis Resistance in Cancer and the Relevance of Metabolic Reprogramming. Front. Oncol. 2021, 11, 626577. [Google Scholar] [CrossRef]
- Kim, Y.N.; Koo, K.H.; Sung, J.Y.; Yun, U.J.; Kim, H. Anoikis resistance: An essential prerequisite for tumor metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef]
- Raeisi, M.; Zehtabi, M.; Velaei, K.; Fayyazpour, P.; Aghaei, N.; Mehdizadeh, A. Anoikis in cancer: The role of lipid signaling. Cell Biol. Int. 2022. [Google Scholar] [CrossRef]
- Slattum, G.M.; Rosenblatt, J. Tumour cell invasion: An emerging role for basal epithelial cell extrusion. Nat. Rev. Cancer 2014, 14, 495–501. [Google Scholar] [CrossRef]
- Alanko, J.; Ivaska, J. Endosomes: Emerging Platforms for Integrin-Mediated FAK Signalling. Trends Cell Biol. 2016, 26, 391–398. [Google Scholar] [CrossRef]
- Li, K.; Zhao, G.; Ao, J.; Gong, D.; Zhang, J.; Chen, Y.; Li, J.; Huang, L.; Xiang, R.; Hu, J.; et al. ZNF32 induces anoikis resistance through maintaining redox homeostasis and activating Src/FAK signaling in hepatocellular carcinoma. Cancer Lett. 2019, 442, 271–278. [Google Scholar] [CrossRef]
- Mason, J.A.; Davison-Versagli, C.A.; Leliaert, A.K.; Pape, D.J.; McCallister, C.; Zuo, J.; Durbin, S.M.; Buchheit, C.L.; Zhang, S.; Schafer, Z.T. Oncogenic Ras differentially regulates metabolism and anoikis in extracellular matrix-detached cells. Cell Death Differ. 2016, 23, 1271–1282. [Google Scholar] [CrossRef]
- Song, J.; Liu, Y.; Liu, F.; Zhang, L.; Li, G.; Yuan, C.; Yu, C.; Lu, X.; Liu, Q.; Chen, X.; et al. The 14-3-3σ protein promotes HCC anoikis resistance by inhibiting EGFR degradation and thereby activating the EGFR-dependent ERK1/2 signaling pathway. Theranostics 2021, 11, 996–1015. [Google Scholar] [CrossRef]
- Sharma, R.; Gogoi, G.; Saikia, S.; Sharma, A.; Kalita, D.J.; Sarma, A.; Limaye, A.M.; Gaur, M.K.; Bhattacharyya, J.; Jaganathan, B.G. BMP4 enhances anoikis resistance and chemoresistance of breast cancer cells through canonical BMP signaling. J. Cell Commun. Signal 2022, 16, 191–205. [Google Scholar] [CrossRef]
- Moro, L.; Arbini, A.A.; Yao, J.L.; di Sant’Agnese, P.A.; Marra, E.; Greco, M. Mitochondrial DNA depletion in prostate epithelial cells promotes anoikis resistance and invasion through activation of PI3K/Akt2. Cell Death Differ. 2009, 16, 571–583. [Google Scholar] [CrossRef]
- Wang, L.N.; Zhang, Z.T.; Wang, L.; Wei, H.X.; Zhang, T.; Zhang, L.M.; Lin, H.; Zhang, H.; Wang, S.Q. TGF-β1/SH2B3 axis regulates anoikis resistance and EMT of lung cancer cells by modulating JAK2/STAT3 and SHP2/Grb2 signaling pathways. Cell Death Dis. 2022, 13, 472. [Google Scholar] [CrossRef]
- Luey, B.C.; May, F.E. Insulin-like growth factors are essential to prevent anoikis in oestrogen-responsive breast cancer cells: Importance of the type I IGF receptor and PI3-kinase/Akt pathway. Mol. Cancer 2016, 15, 8. [Google Scholar] [CrossRef]
- Patidar, A.; Selvaraj, S.; Sarode, A.; Chauhan, P.; Chattopadhyay, D.; Saha, B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 2018, 104, 114–123. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rivandi, M.; Abedini, S.; Pasdar, A.; Sahebkar, A. Regulators and mechanisms of anoikis in triple-negative breast cancer (TNBC): A review. Crit. Rev. Oncol. Hematol. 2019, 140, 17–27. [Google Scholar] [CrossRef]
- Zhu, Z.; Fang, C.; Xu, H.; Yuan, L.; Du, Y.; Ni, Y.; Xu, Y.; Shao, A.; Zhang, A.; Lou, M. Anoikis resistance in diffuse glioma: The potential therapeutic targets in the future. Front. Oncol. 2022, 12, 976557. [Google Scholar] [CrossRef]
- Sakamoto, S.; Kyprianou, N. Targeting anoikis resistance in prostate cancer metastasis. Mol. Asp. Med. 2010, 31, 205–214. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Multiple roles of glyoxalase 1-mediated suppression of methylglyoxal glycation in cancer biology-Involvement in tumour suppression, tumour growth, multidrug resistance and target for chemotherapy. Semin. Cancer Biol. 2018, 49, 83–93. [Google Scholar] [CrossRef]
- Panchal, N.K.; Evan Prince, S. The NEK family of serine/threonine kinases as a biomarker for cancer. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Z. HBV-Induced Immune Imbalance in the Development of HCC. Front. Immunol. 2019, 10, 2048. [Google Scholar] [CrossRef]
- Lin, L.; Chen, S.; Wang, H.; Gao, B.; Kallakury, B.; Bhuvaneshwar, K.; Cahn, K.; Gusev, Y.; Wang, X.; Wu, Y.; et al. SPTBN1 inhibits inflammatory responses and hepatocarcinogenesis via the stabilization of SOCS1 and downregulation of p65 in hepatocellular carcinoma. Theranostics 2021, 11, 4232–4250. [Google Scholar] [CrossRef]
- Saviano, A.; Roehlen, N.; Virzì, A.; Roca Suarez, A.A.; Hoshida, Y.; Lupberger, J.; Baumert, T.F. Stromal and Immune Drivers of Hepatocarcinogenesis. In Hepatocellular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Humana Press Copyright 2019; Springer Nature Switzerland AG.: Cham, Switzerland, 2019; pp. 317–331. [Google Scholar]
- Ormandy, L.A.; Farber, A.; Cantz, T.; Petrykowska, S.; Wedemeyer, H.; Horning, M.; Lehner, F.; Manns, M.P.; Korangy, F.; Greten, T.F. Direct ex vivo analysis of dendritic cells in patients with hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 3275–3282. [Google Scholar] [CrossRef]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Zhou, Z.; Deng, H.; Yan, W.; Luo, M.; Tu, W.; Xia, Y.; He, J.; Han, P.; Fu, Y.; Tian, D. AEG-1 promotes anoikis resistance and orientation chemotaxis in hepatocellular carcinoma cells. PLoS ONE 2014, 9, e100372. [Google Scholar] [CrossRef]
- Dadsena, S.; King, L.E.; García-Sáez, A.J. Apoptosis regulation at the mitochondria membrane level. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183716. [Google Scholar] [CrossRef]
- Alavi, M.V. Targeted OMA1 therapies for cancer. Int. J. Cancer 2019, 145, 2330–2341. [Google Scholar] [CrossRef]
- Ramesh, P.; Medema, J.P. BCL-2 family deregulation in colorectal cancer: Potential for BH3 mimetics in therapy. Apoptosis 2020, 25, 305–320. [Google Scholar] [CrossRef]
- Park, D.; Anisuzzaman, A.S.M.; Magis, A.T.; Chen, G.; Xie, M.; Zhang, G.; Behera, M.; Sica, G.L.; Ramalingam, S.S.; Owonikoko, T.K.; et al. Discovery of Small Molecule Bak Activator for Lung Cancer Therapy. Theranostics 2021, 11, 8500–8516. [Google Scholar] [CrossRef]
- Han, J.; Chen, C.; Wang, C.; Qin, N.; Huang, M.; Ma, Z.; Zhu, M.; Dai, J.; Jiang, Y.; Ma, H.; et al. Transcriptome-wide association study for persistent hepatitis B virus infection and related hepatocellular carcinoma. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 2117–2127. [Google Scholar] [CrossRef]
- Lai, M.; Liu, Q.; Chen, W.; Qi, X.; Yang, J.; Jiang, L.; Yuan, M.; Liu, Z.; He, Q.; Cao, J.; et al. Identification and Validation of Two Heterogeneous Molecular Subtypes and a Prognosis Predictive Model for Hepatocellular Carcinoma Based on Pyroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 8346816. [Google Scholar] [CrossRef]
- Wazir, U.; Orakzai, M.M.; Khanzada, Z.S.; Jiang, W.G.; Sharma, A.K.; Kasem, A.; Mokbel, K. The role of death-associated protein 3 in apoptosis, anoikis and human cancer. Cancer Cell Int. 2015, 15, 39. [Google Scholar] [CrossRef] [Green Version]
- Li, H.M.; Fujikura, D.; Harada, T.; Uehara, J.; Kawai, T.; Akira, S.; Reed, J.C.; Iwai, A.; Miyazaki, T. IPS-1 is crucial for DAP3-mediated anoikis induction by caspase-8 activation. Cell Death Differ. 2009, 16, 1615–1621. [Google Scholar] [CrossRef]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Kaczmarek, E.; Ponce, F.; Coons, S.W.; Giese, A.; Seiler, R.W.; Berens, M.E. Death-associated protein 3 (Dap-3) is overexpressed in invasive glioblastoma cells in vivo and in glioma cell lines with induced motility phenotype in vitro. Clin. Cancer Res. 2001, 7, 2480–2489. [Google Scholar]
- Huang, T.; He, W.Y. Pan-Cancer Analysis, Reveals COVID-19-Related BSG as a Novel Marker for Treatment and Identification of Multiple Human Cancers. Front. Cell Dev. Biol. 2022, 10, 876180. [Google Scholar] [CrossRef]
- Ma, Y.; Sun, W.; Zhang, Q.; Gao, B.; Cai, W.; Liu, Q.; Liao, J.; Wang, X. lncRNA BSG-AS1 is hypoxia-responsive and promotes hepatocellular carcinoma by enhancing BSG mRNA stability. Biochem. Biophys. Res. Commun. 2021, 566, 101–107. [Google Scholar] [CrossRef]
- Yang, L.; Deng, W.L.; Zhao, B.G.; Xu, Y.; Wang, X.W.; Fang, Y.; Xiao, H.J. FOXO3-induced lncRNA LOC554202 contributes to hepatocellular carcinoma progression via the miR-485-5p/BSG axis. Cancer Gene Ther. 2022, 29, 326–340. [Google Scholar] [CrossRef]
- Nallasamy, P.; Nimmakayala, R.K.; Karmakar, S.; Leon, F.; Seshacharyulu, P.; Lakshmanan, I.; Rachagani, S.; Mallya, K.; Zhang, C.; Ly, Q.P.; et al. Pancreatic Tumor Microenvironment Factor Promotes Cancer Stemness via SPP1-CD44 Axis. Gastroenterology 2021, 161, 1998–2013.e7. [Google Scholar] [CrossRef]
- Pang, X.; Zhang, J.; He, X.; Gu, Y.; Qian, B.Z.; Xie, R.; Yu, W.; Zhang, X.; Li, T.; Shi, X.; et al. SPP1 Promotes Enzalutamide Resistance and Epithelial-Mesenchymal-Transition Activation in Castration-Resistant Prostate Cancer via PI3K/AKT and ERK1/2 Pathways. Oxidative Med. Cell. Longev. 2021, 2021, 5806602. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Z.; Xia, Y.; Cai, Z.; Wang, C.; Shen, Y.; Liu, R.; Qin, H.; Jia, J.; Yuan, G. Potential biomarkers in the fibrosis progression of nonalcoholic steatohepatitis (NASH). J. Endocrinol. Investig. 2022, 45, 1379–1392. [Google Scholar] [CrossRef]
- Wu, X.; Sui, Z.; Zhang, H.; Wang, Y.; Yu, Z. Integrated Analysis of lncRNA-Mediated ceRNA Network in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 554759. [Google Scholar] [CrossRef]
- Nagano-Matsuo, A.; Inoue, S.; Koshino, A.; Ota, A.; Nakao, K.; Komura, M.; Kato, H.; Naiki-Ito, A.; Watanabe, K.; Nagayasu, Y.; et al. PBK expression predicts favorable survival in colorectal cancer patients. Virchows Arch. 2021, 479, 277–284. [Google Scholar] [CrossRef]
- Kwon, C.H.; Park, H.J.; Choi, Y.R.; Kim, A.; Kim, H.W.; Choi, J.H.; Hwang, C.S.; Lee, S.J.; Choi, C.I.; Jeon, T.Y.; et al. PSMB8 and PBK as potential gastric cancer subtype-specific biomarkers associated with prognosis. Oncotarget 2016, 7, 21454–21468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.X.; Zhong, S.; He, L.; Jia, X.J.; Tang, H.; Cheng, S.T.; Ren, J.H.; Yu, H.B.; Zhou, L.; Zhou, H.Z.; et al. PBK overexpression promotes metastasis of hepatocellular carcinoma via activating ETV4-uPAR signaling pathway. Cancer Lett. 2019, 452, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Pan, Y.H.; Cao, Y.; Fu, J.; Yang, X.; Zhang, M.F.; Tian, Q.H. PDZ binding kinase, regulated by FoxM1, enhances malignant phenotype via activation of β-Catenin signaling in hepatocellular carcinoma. Oncotarget 2017, 8, 47195–47205. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Huang, W.; Ouyang, J.; Wang, J.; Xie, Z. Identification of Anoikis-Related Subgroups and Prognosis Model in Liver Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 2862. https://doi.org/10.3390/ijms24032862
Chen Y, Huang W, Ouyang J, Wang J, Xie Z. Identification of Anoikis-Related Subgroups and Prognosis Model in Liver Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(3):2862. https://doi.org/10.3390/ijms24032862
Chicago/Turabian StyleChen, Yutong, Weiran Huang, Jian Ouyang, Jingxiang Wang, and Zhengwei Xie. 2023. "Identification of Anoikis-Related Subgroups and Prognosis Model in Liver Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 3: 2862. https://doi.org/10.3390/ijms24032862