
Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
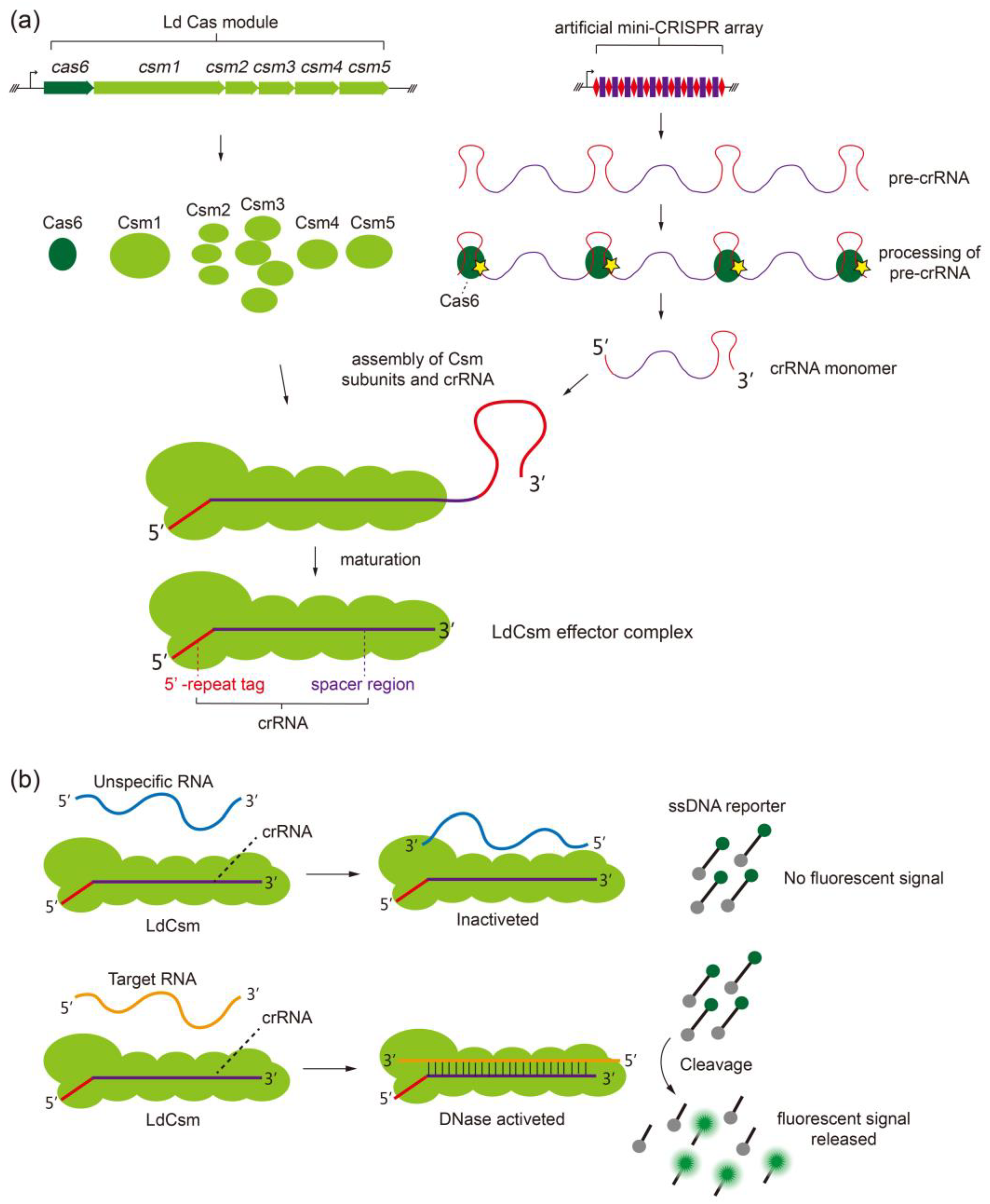
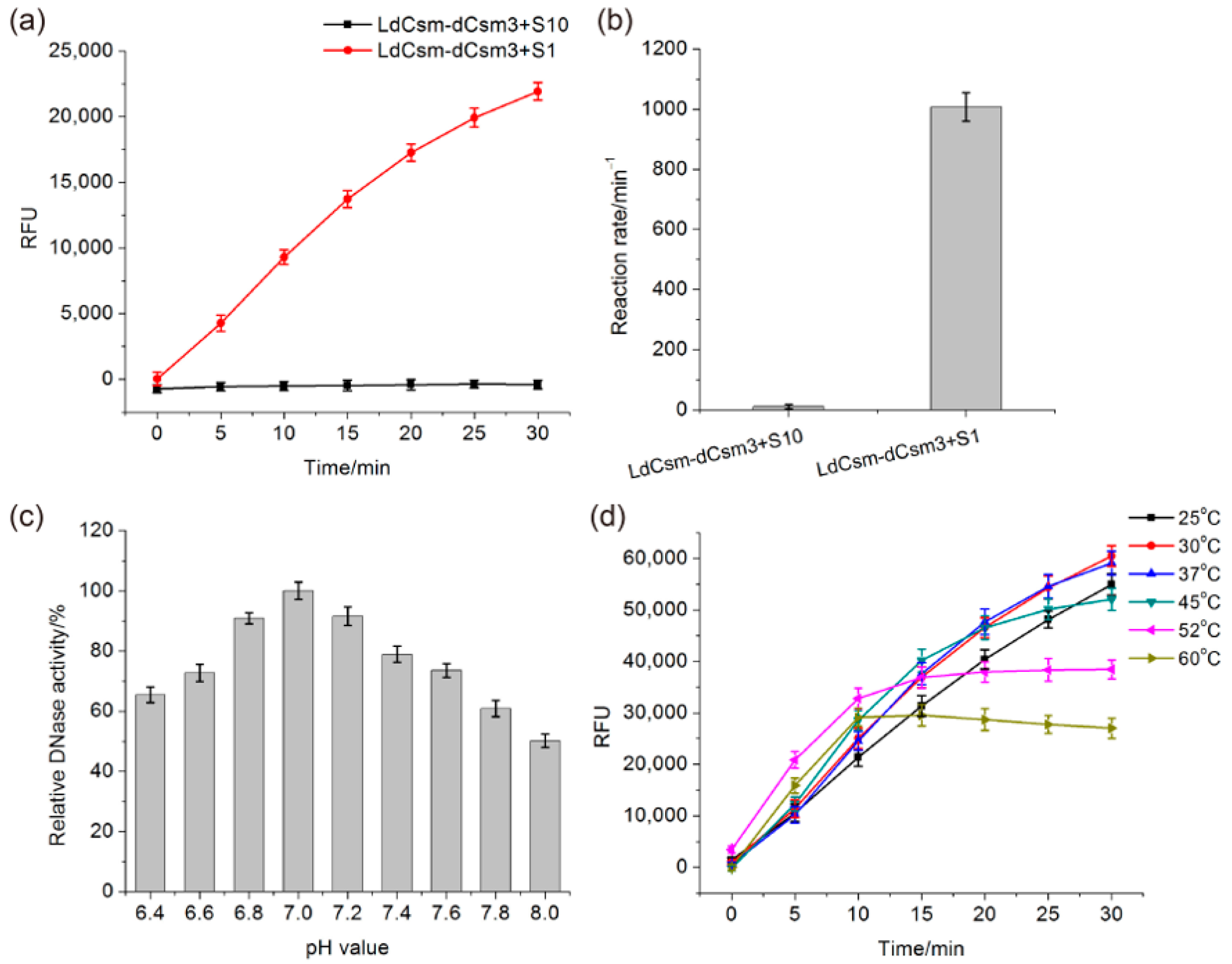
2.1. Developing the LdCsm RNA Detection Platform
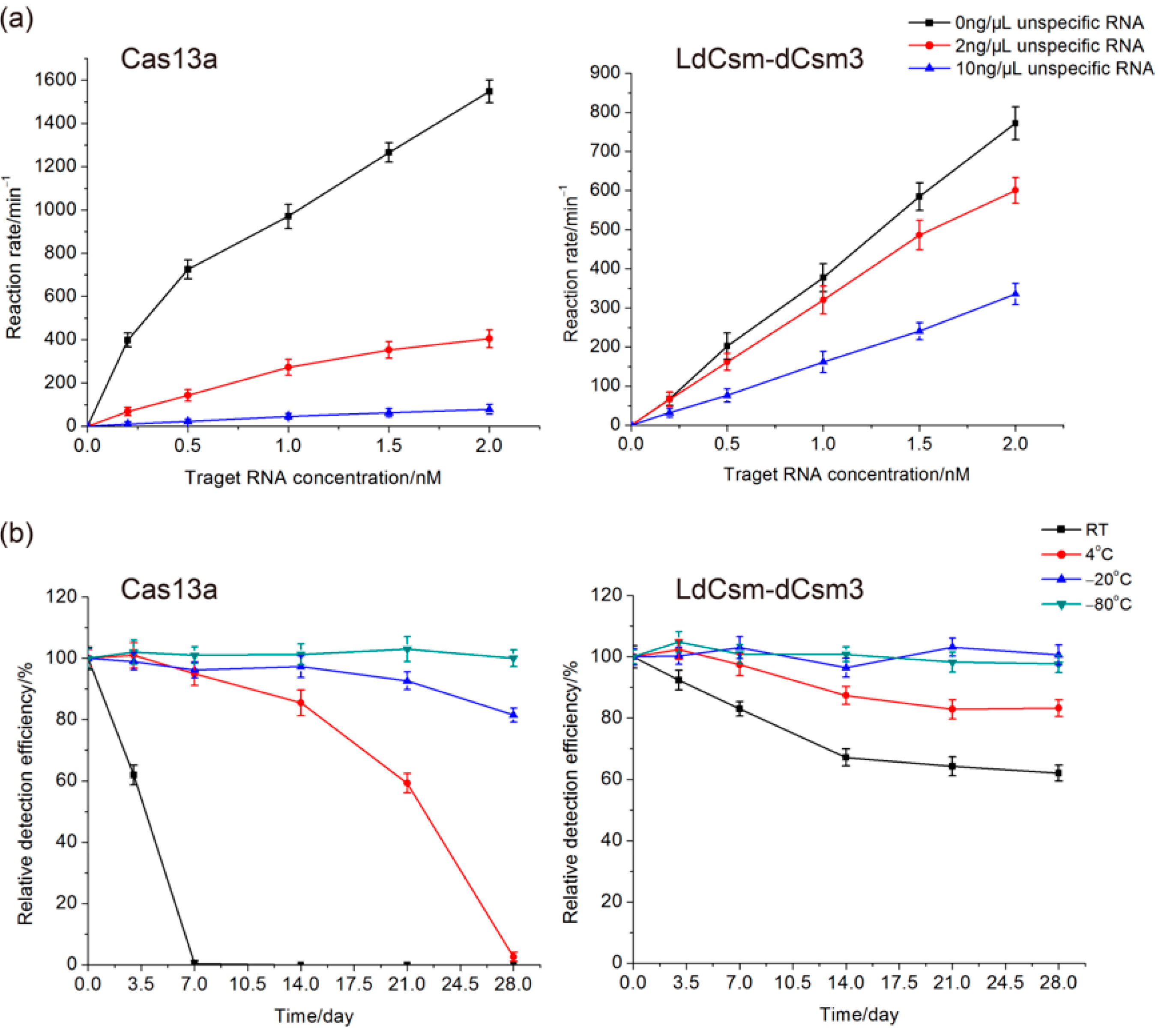
2.2. Comparison of LdCsm and Cas13a Detection Systems
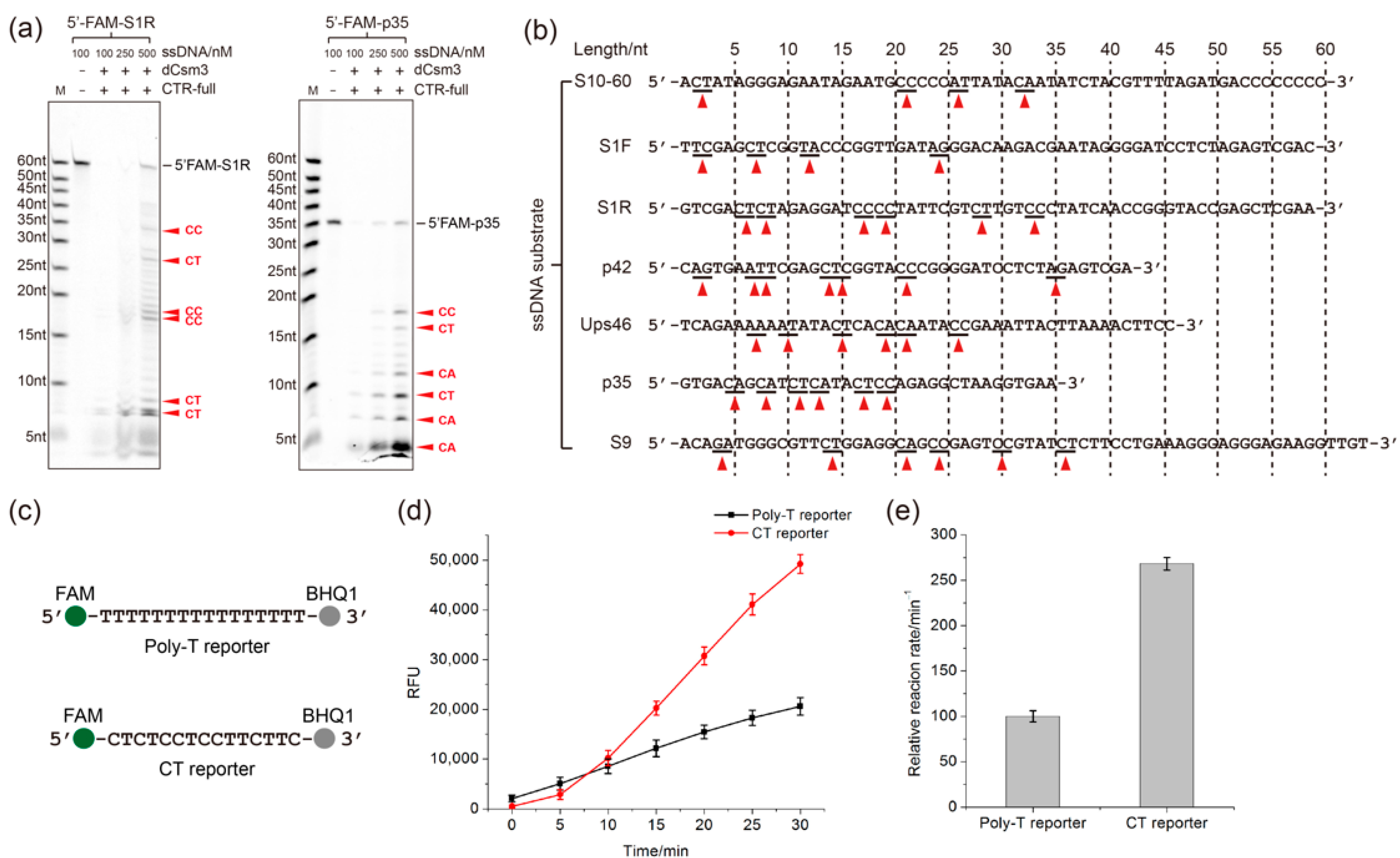
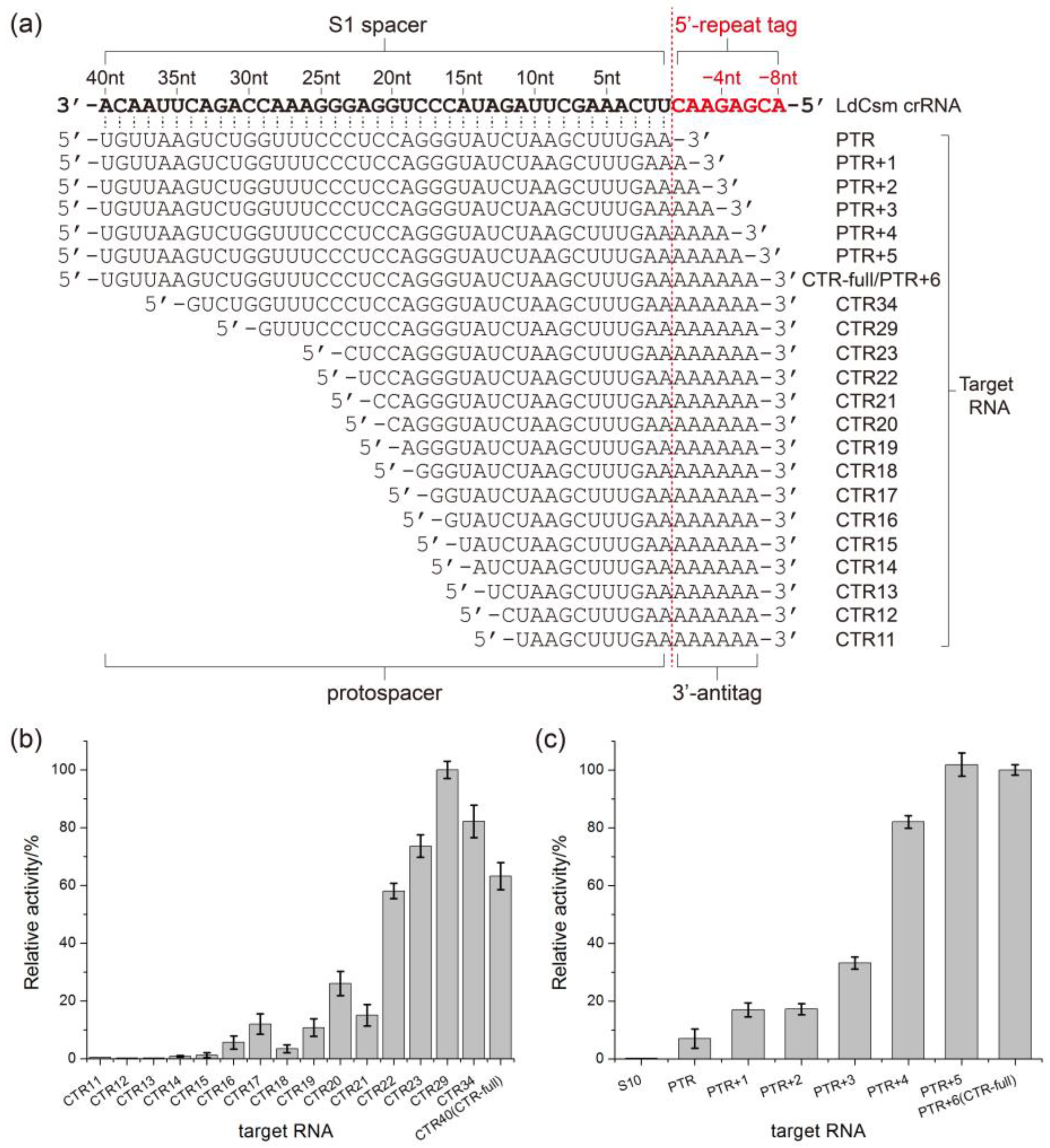
2.3. Determination of the Length Requirement of Activator RNA in the LdCsm-dCsm3 Detection Platform
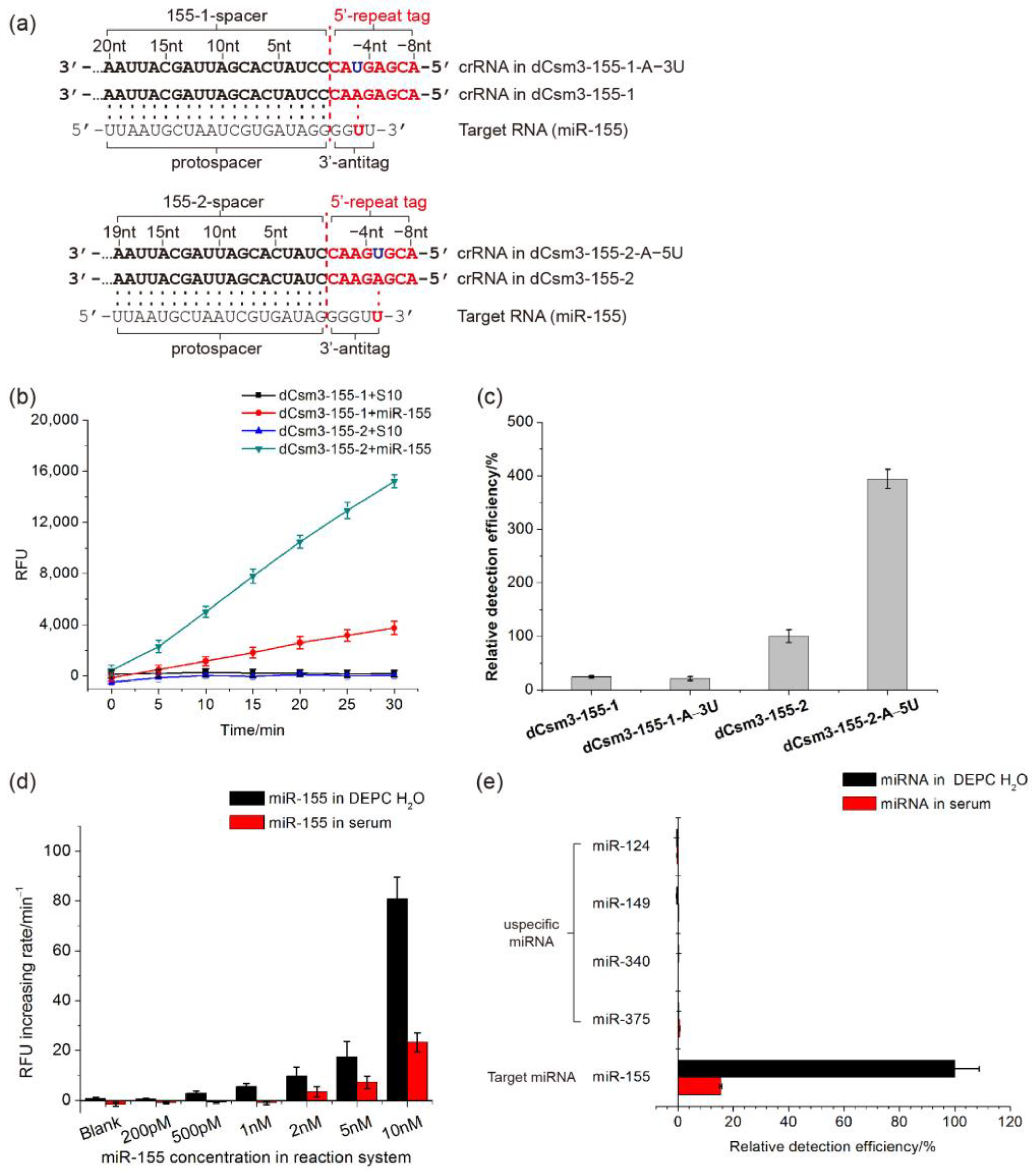
2.4. Developing the LdCsm-dCsm3-Based microRNA Detection System
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Construction of Artificial Mini-CRISPR Plasmids Carrying Different Spacers or Repeats
4.3. Purification of LdCsm Effector Complexes from E. coli
4.4. Collateral Nucleic Acid Cleavage Assay
4.5. Evaluation of Fluorescence DNA Reporter Cleavage Assay/RNA Detection Reaction
4.6. RNA Detection Reaction Using Cas13a
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Prim. 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Touchette, N.A.; Folkers, G.K. Emerging infectious diseases: A 10-year perspective from the National Institute of Allergy and Infectious Diseases. Emerg. Infect. Dis. 2005, 11, 519–525. [Google Scholar] [CrossRef]
- Masmejan, S.; Musso, D.; Vouga, M.; Pomar, L.; Dashraath, P.; Stojanov, M.; Panchaud, A.; Baud, D. Zika Virus. Pathogens 2020, 9, 898. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.T.; Crozier, I.; Fischer, W.A., 2nd; Hewlett, A.; Kraft, C.S.; Vega, M.A.; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola virus disease. Nat. Rev. Dis. Prim. 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.Q.; Peng, H.J. Characteristics of and Public Health Responses to the Coronavirus Disease 2019 Outbreak in China. J. Clin. Med. 2020, 9, 575. [Google Scholar] [CrossRef]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer Res. 2020, 126, 2225–2249. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tutar, L.; Özgür, A.; Tutar, Y. Involvement of miRNAs and Pseudogenes in Cancer. Methods Mol. Biol. 2018, 1699, 45–66. [Google Scholar]
- Huang, R.; Yu, H.; Zhong, X. Identification of Novel CircRNA-miRNA-mRNA Regulatory Network and Its Prognostic Prediction in Breast Cancer. Evid.-Based Complement. Altern. Med. Ecam 2021, 2021, 2916398. [Google Scholar] [CrossRef]
- Chen, L.; Shan, G. CircRNA in cancer: Fundamental mechanism and clinical potential. Cancer Lett. 2021, 505, 49–57. [Google Scholar] [CrossRef]
- Gurukumar, K.R.; Priyadarshini, D.; Patil, J.A.; Bhagat, A.; Singh, A.; Shah, P.S.; Cecilia, D. Development of real time PCR for detection and quantitation of Dengue Viruses. Virol. J. 2009, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watzinger, F.; Ebner, K.; Lion, T. Detection and monitoring of virus infections by real-time PCR. Mol. Asp. Med. 2006, 27, 254–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tan, R.; Wong, L.; Fekete, R.; Halsey, J. Quantitation of microRNAs by real-time RT-qPCR. Methods Mol. Biol. 2011, 687, 113–134. [Google Scholar] [PubMed]
- Abd El Wahed, A.; Sanabani, S.S.; Faye, O.; Pessôa, R.; Patriota, J.V.; Giorgi, R.R.; Patel, P.; Böhlken-Fascher, S.; Landt, O.; Niedrig, M.; et al. Rapid Molecular Detection of Zika Virus in Acute-Phase Urine Samples Using the Recombinase Polymerase Amplification Assay. PLoS Curr. 2017, 9. [Google Scholar] [CrossRef]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Herbst, K.; Boerner, K.; Meurer, M.; Kremer, L.P.; Kirrmaier, D.; Freistaedter, A.; Papagiannidis, D.; Galmozzi, C.; Stanifer, M.L.; et al. A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 2020, 12, eabc7075. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, G.; Buss, J.; Barry, A.J.; Patton, G.C.; Tanner, N.A. Enhancing colorimetric loop-mediated isothermal amplification speed and sensitivity with guanidine chloride. BioTechniques 2020, 69, 178–185. [Google Scholar] [CrossRef]
- Lu, W.; Wang, Y.; Song, S.; Chen, C.; Yao, B.; Wang, M. A fishhook probe-based rolling circle amplification (FP-RCA) assay for efficient isolation and detection of microRNA without total RNA extraction. Analyst 2018, 143, 5046–5053. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Bi, Y.; Sun, M.; Bai, J.; Zhou, M. Ultrasensitive electrochemiluminescence biosensing platform for miRNA-21 and MUC1 detection based on dual catalytic hairpin assembly. Anal. Chim. Acta 2020, 1105, 87–94. [Google Scholar] [CrossRef]
- Gu, J.; Qiao, Z.; He, X.; Yu, Y.; Lei, Y.; Tang, J.; Shi, H.; He, D.; Wang, K. Enzyme-free amplified detection of miRNA based on target-catalyzed hairpin assembly and DNA-stabilized fluorescent silver nanoclusters. Analyst 2020, 145, 5194–5199. [Google Scholar] [CrossRef]
- Ning, L.; Cheng, H.; Yu, F.; Zhou, Y.; Xie, Y. Construction of simple and sensitive pancreatitis related microRNA detection strategy via self-priming triggered cascade signal amplification. Anal. Bioanal. Chem. 2022, 414, 5837–5844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q.; Weng, X.; Du, Y.; Zhou, X. NEase-based amplification for detection of miRNA, multiple miRNAs and circRNA. Anal. Chim. Acta 2021, 1145, 52–58. [Google Scholar] [CrossRef]
- Zhao, G.; Yan, X.; Zhang, Y.; Deng, J.; Liang, X. Sensitive detection of MiRNA and CircRNA through DSN enzyme cooperating NEase assisted dual signal amplification. Anal. Biochem. 2022, 654, 114744. [Google Scholar] [CrossRef] [PubMed]
- Meagher, R.J.; Priye, A.; Light, Y.K.; Huang, C.; Wang, E. Impact of primer dimers and self-amplifying hairpins on reverse transcription loop-mediated isothermal amplification detection of viral RNA. Analyst 2018, 143, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Rolando, J.C.; Jue, E.; Barlow, J.T.; Ismagilov, R.F. Real-time kinetics and high-resolution melt curves in single-molecule digital LAMP to differentiate and study specific and non-specific amplification. Nucleic Acids Res. 2020, 48, e42. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Mason, M.G.; Botella, J.R. Evaluation and improvement of isothermal amplification methods for point-of-need plant disease diagnostics. PloS ONE 2020, 15, e0235216. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Mohanraju, P.; Makarova, K.S.; Zetsche, B.; Zhang, F.; Koonin, E.V.; van der Oost, J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016, 353, aad5147. [Google Scholar] [CrossRef]
- Wright, A.V.; Nunez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, Z.; She, Q.; Han, W. The abortive infection functions of CRISPR-Cas and Argonaute. Trends Microbiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Samai, P.; Pyenson, N.; Jiang, W.; Goldberg, G.W.; Hatoum-Aslan, A.; Marraffini, L.A. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 2015, 161, 1164–1174. [Google Scholar] [CrossRef]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef]
- Santiago-Frangos, A.; Hall, L.N.; Nemudraia, A.; Nemudryi, A.; Krishna, P.; Wiegand, T.; Wilkinson, R.A.; Snyder, D.T.; Hedges, J.F.; Cicha, C.; et al. Intrinsic signal amplification by type III CRISPR-Cas systems provides a sequence-specific SARS-CoV-2 diagnostic. Cell Rep. Med. 2021, 2, 100319. [Google Scholar] [CrossRef]
- Steens, J.A.; Zhu, Y.; Taylor, D.W.; Bravo, J.P.K.; Prinsen, S.H.P.; Schoen, C.D.; Keijser, B.J.F.; Ossendrijver, M.; Hofstra, L.M.; Brouns, S.J.J.; et al. SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat. Commun. 2021, 12, 5033. [Google Scholar] [CrossRef]
- Sridhara, S.; Goswami, H.N.; Whyms, C.; Dennis, J.H.; Li, H. Virus detection via programmable Type III-A CRISPR-Cas systems. Nat. Commun. 2021, 12, 5653. [Google Scholar] [CrossRef]
- Grüschow, S.; Adamson, C.S.; White, M.F. Specificity and sensitivity of an RNA targeting type III CRISPR complex coupled with a NucC endonuclease effector. Nucleic Acids Res. 2021, 49, 13122–13134. [Google Scholar] [CrossRef]
- Peng, S.; Tan, Z.; Chen, S.; Lei, C.; Nie, Z. Integrating CRISPR-Cas12a with a DNA circuit as a generic sensing platform for amplified detection of microRNA. Chem. Sci. 2020, 11, 7362–7368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, Z.; Lu, J.; Ren, X.; Ma, Y. Ultrasensitive visual detection of miRNA-143 using a CRISPR/Cas12a-based platform coupled with hyperbranched rolling circle amplification. Talanta 2023, 251, 123784. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, J.; He, N.; Zhang, M.; Wu, L.; Chen, X.; Zhu, J.; Ran, F.; Chen, Q.; Zhang, H. CRISPR/Cas12a Coupling with Magnetic Nanoparticles and Cascaded Strand Displacement Reaction for Ultrasensitive Fluorescence Determination of Exosomal miR-21. Molecules 2022, 27, 5338. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.Y.; Zhao, H.L.; Wang, T.; Chen, P.R.; Yin, B.C.; Ye, B.C. A programmable and sensitive CRISPR/Cas12a-based MicroRNA detection platform combined with hybridization chain reaction. Biosens. Bioelectron. 2022, 211, 114382. [Google Scholar] [CrossRef]
- Broto, M.; Kaminski, M.M.; Adrianus, C.; Kim, N.; Greensmith, R.; Dissanayake-Perera, S.; Schubert, A.J.; Stevens, M.M.; Tan, X.; Kim, H.; et al. Nanozyme-catalysed CRISPR assay for preamplification-free detection of non-coding RNAs. Nat. Nanotechnol. 2022, 17, 1120–1126. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, C.; Liu, G.; Zhao, X.; Qian, Q.; Li, S.; Mi, X. Tetrahedral DNA framework based CRISPR electrochemical biosensor for amplification-free miRNA detection. Biosens. Bioelectron. 2022, 217, 114671. [Google Scholar] [CrossRef]
- Lin, J.; Feng, M.; Zhang, H.; She, Q. Characterization of a novel type III CRISPR-Cas effector provides new insights into the allosteric activation and suppression of the Cas10 DNase. Cell Discovry 2020, 6, 29. [Google Scholar] [CrossRef]
- Lin, J.; Shen, Y.; Ni, J.; She, Q. A type III-A CRISPR-Cas system mediates co-transcriptional DNA cleavage at the transcriptional bubbles in close proximity to active effectors. Nucleic Acids Res. 2021, 49, 7628–7643. [Google Scholar] [CrossRef]
- Han, W.; Li, Y.; Deng, L.; Feng, M.; Peng, W.; Hallstrom, S.; Zhang, J.; Peng, N.; Liang, Y.X.; White, M.F.; et al. A type III-B CRISPR-Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 2017, 45, 1983–1993. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.L.; Paul, L.M.; Parham, L.A.; et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Catela Ivkovic, T.; Voss, G.; Cornella, H.; Ceder, Y. microRNAs as cancer therapeutics: A step closer to clinical application. Cancer Lett. 2017, 407, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Van Roosbroeck, K. miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018, 37, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.M.; Nuzzo, S.; Condorelli, G.; Salvatore, M.; Incoronato, M. Prognostic and Clinicopathological Significance of MiR-155 in Breast Cancer: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 5834. [Google Scholar] [CrossRef] [PubMed]
- Sueta, A.; Yamamoto, Y.; Tomiguchi, M.; Takeshita, T.; Yamamoto-Ibusuki, M.; Iwase, H. Differential expression of exosomal miRNAs between breast cancer patients with and without recurrence. Oncotarget 2017, 8, 69934–69944. [Google Scholar] [CrossRef]
- Zhang, M.; Bai, X.; Zeng, X.; Liu, J.; Liu, F.; Zhang, Z. circRNA-miRNA-mRNA in breast cancer. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 523, 120–130. [Google Scholar] [CrossRef]
- Zhang, X.; An, X. Adaptation by Type III CRISPR-Cas Systems: Breakthrough Findings and Open Questions. Front. Microbiol. 2022, 13, 876174. [Google Scholar] [CrossRef]
- Kazlauskiene, M.; Kostiuk, G.; Siksnys, V.; Tamulaitis, G.; Venclovas, Č. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 2017, 357, 605–609. [Google Scholar] [CrossRef]
- Niewoehner, O.; Garcia-Doval, C.; Rostøl, J.T.; Berk, C.; Schwede, F.; Bigler, L.; Hall, J.; Marraffini, L.A.; Jinek, M. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 2017, 548, 543–548. [Google Scholar] [CrossRef]
- Han, W.; Stella, S.; Zhang, Y.; Guo, T.; Sulek, K.; Peng-Lundgren, L.; Montoya, G.; She, Q. A Type III-B Cmr effector complex catalyzes the synthesis of cyclic oligoadenylate second messengers by cooperative substrate binding. Nucleic Acids Res. 2018, 46, 10319–10330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, S.A.; Zhu, W.; Graham, S.; Rambo, R.; White, M.F.; Gloster, T.M. Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. Nat. Commun. 2020, 11, 500. [Google Scholar] [CrossRef] [PubMed]
- Rostøl, J.T.; Xie, W.; Kuryavyi, V.; Maguin, P.; Kao, K.; Froom, R.; Patel, D.J.; Marraffini, L.A. The Card1 nuclease provides defence during type III CRISPR immunity. Nature 2021, 590, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; McQuarrie, S.; Grüschow, S.; McMahon, S.A.; Graham, S.; Gloster, T.M.; White, M.F. The CRISPR ancillary effector Can2 is a dual-specificity nuclease potentiating type III CRISPR defence. Nucleic Acids Res. 2021, 49, 2777–2789. [Google Scholar] [CrossRef]
- Guo, Z.; Tan, X.; Yuan, H.; Zhang, L.; Wu, J.; Yang, Z.; Qu, K.; Wan, Y. Bis-enzyme cascade CRISPR-Cas12a platform for miRNA detection. Talanta 2023, 252, 123837. [Google Scholar] [CrossRef]
- Wolfinger, R.D.; Beedanagari, S.; Boitier, E.; Chen, T.; Couttet, P.; Ellinger-Ziegelbauer, H.; Guillemain, G.; Mariet, C.; Mouritzen, P.; O’Lone, R.; et al. Two approaches for estimating the lower limit of quantitation (LLOQ) of microRNA levels assayed as exploratory biomarkers by RT-qPCR. BMC Biotechnol. 2018, 18, 6. [Google Scholar] [CrossRef]
- Iguchi, T.; Niino, N.; Tamai, S.; Sakurai, K.; Mori, K. Absolute Quantification of Plasma MicroRNA Levels in Cynomolgus Monkeys, Using Quantitative Real-time Reverse Transcription PCR. J. Vis. Exp. JoVE 2018, 132, e56850. [Google Scholar] [CrossRef]
- Guo, X.; Tian, T.; Deng, X.; Song, Y.; Zhou, X.; Song, E. CRISPR/Cas13a assisted amplification of magnetic relaxation switching sensing for accurate detection of miRNA-21 in human serum. Anal. Chim. Acta 2022, 1209, 339853. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.; Xu, J.; She, Q. Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection. Int. J. Mol. Sci. 2023, 24, 2857. https://doi.org/10.3390/ijms24032857
Yu Z, Xu J, She Q. Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection. International Journal of Molecular Sciences. 2023; 24(3):2857. https://doi.org/10.3390/ijms24032857
Chicago/Turabian StyleYu, Zhenxiao, Jianan Xu, and Qunxin She. 2023. "Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection" International Journal of Molecular Sciences 24, no. 3: 2857. https://doi.org/10.3390/ijms24032857