
Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge
 , , , and
, , , and
Abstract
:1. Introduction
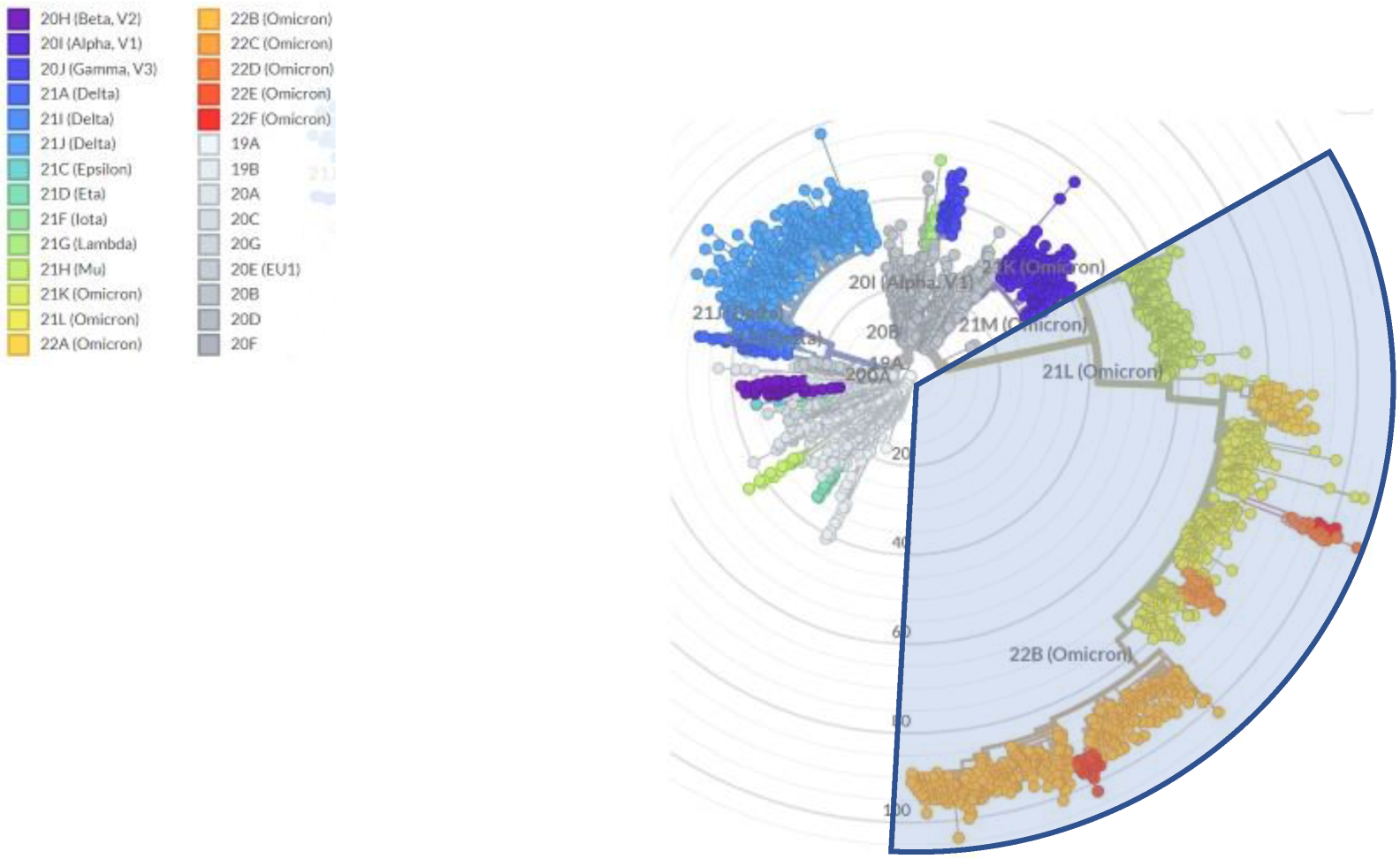
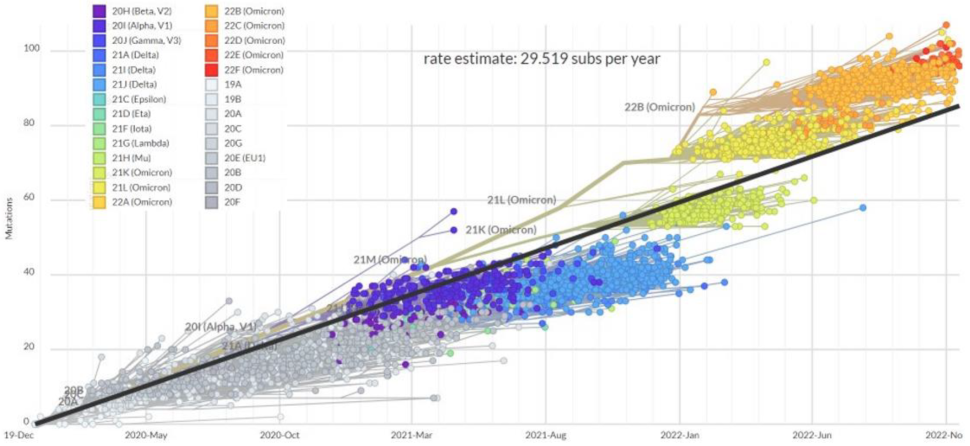
2. Mutation Rates and Mutational Spectra
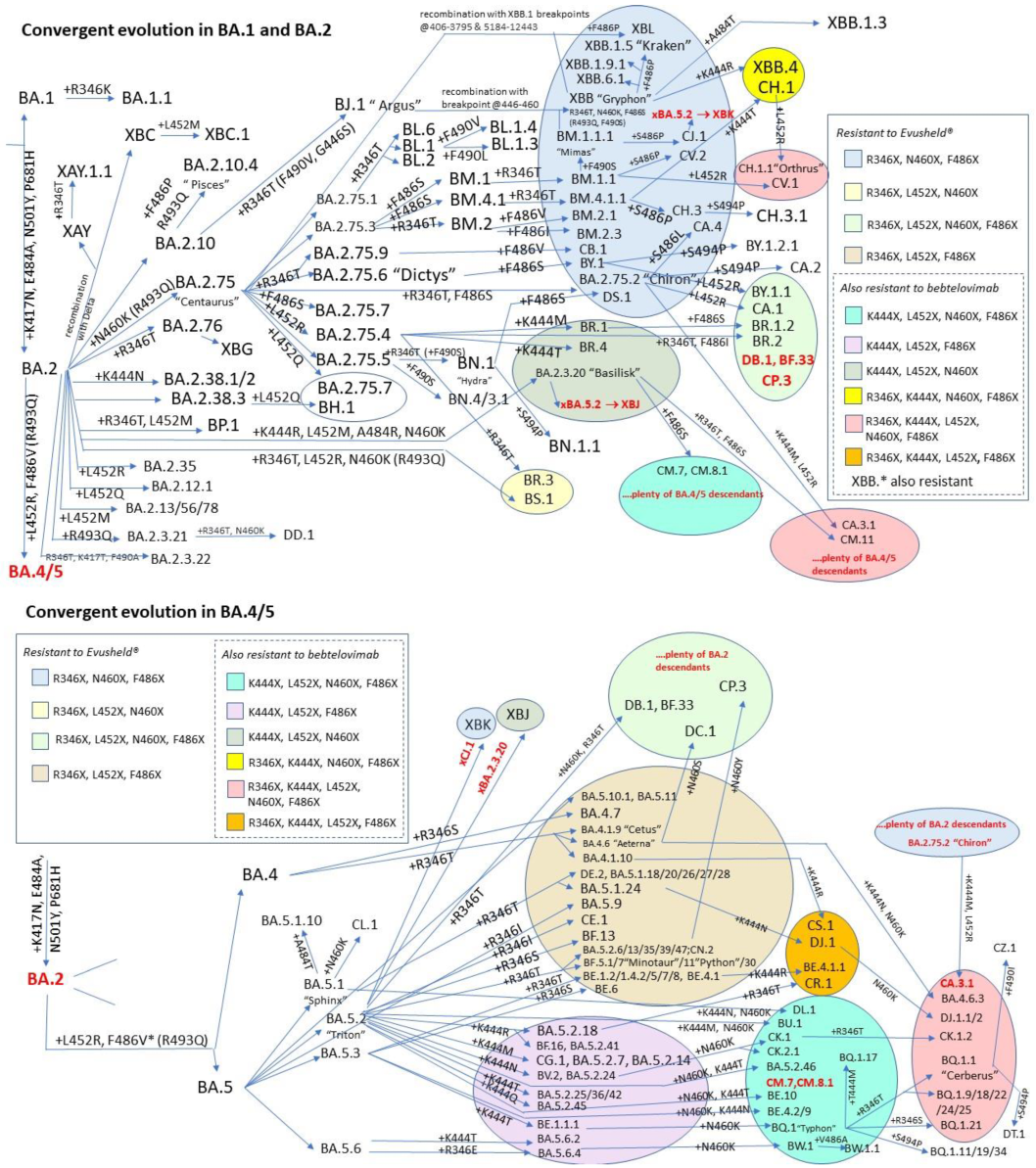
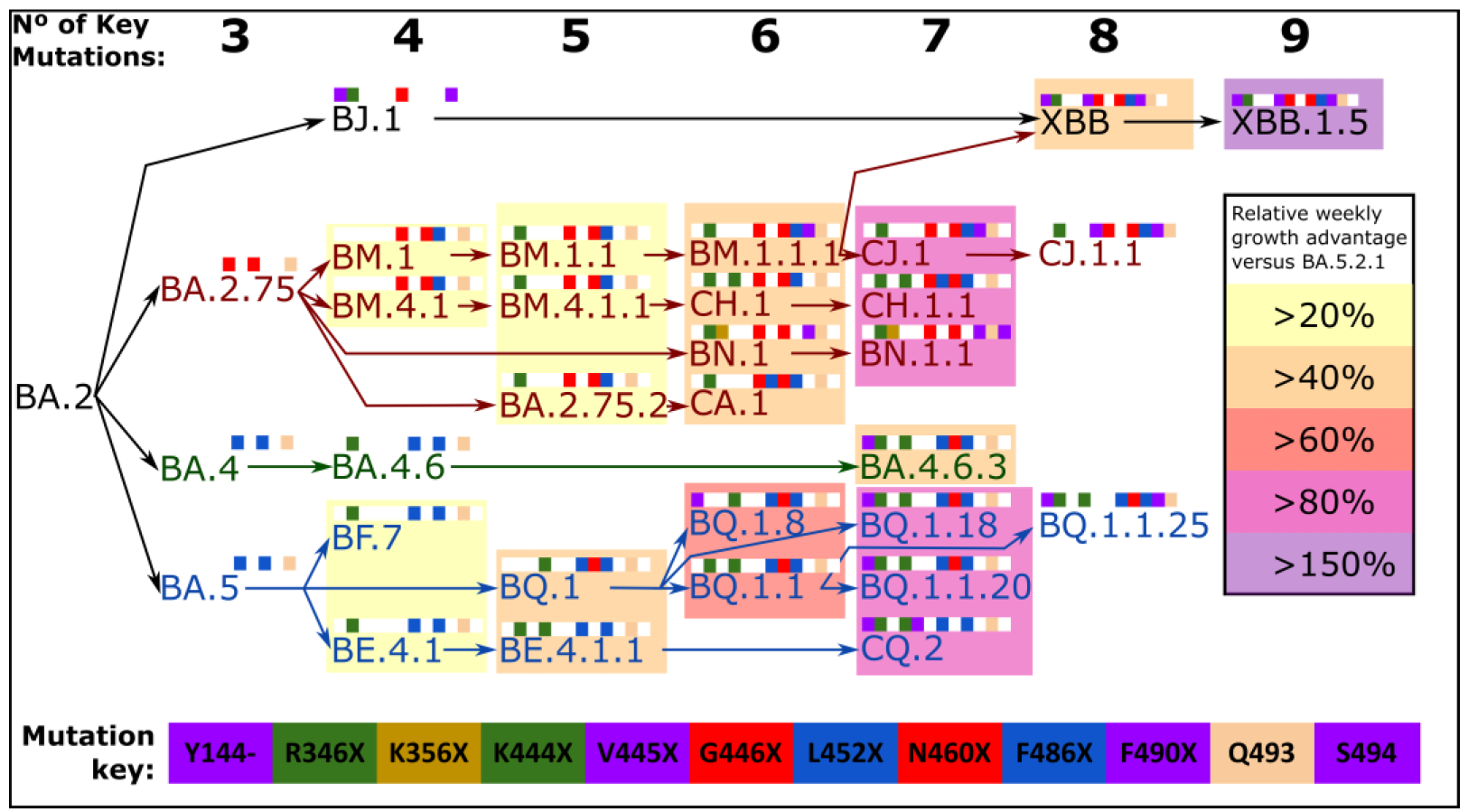
3. Convergent Evolution
4. Escalating Immune Escape
5. ACE2 Affinity Fine Tuning
- BQ.1 already had K444T inherited from BE.1.1.1, but further mutated into 444M in the child BQ.1.1.17
- XBB.1 already had E484A inherited from the BA.2 parent, but further mutated into 484T in the child XBB.1.3
- BA.2.3 already had E484A inherited from the BA.2 parent, but further mutated into 484R in the child BA.2.3.20, which caused an impressive increase in ACE2 affinity (to whom K444R, L452M, and N460K contributed)
- BM.4.1.1 already had F486S inherited from the BM.4.1 parent but further mutated into 486P in CH.3
- BM.1.1.1 already had F486S inherited from the BM.1 parent but further mutated into 486P in the child CJ.1
- XBB.1 already had F486S inherited from the BM.1.1.1 parent but further mutated into 486P in the child XBB.1.5
- BA.2.75.2 already had F486S inherited from the BA.2.75 parent, but further mutated into 486L in the child CA.4
- BA.5.2.1 already had F486V inherited since BA.5, but further mutated to 486I in BF.12
- BW.1 already had F486V inherited from the BA.5 parent, but further mutated into 486S in the child BW.1.1
6. Mutually Exclusive Mutations
7. Epistasis
8. Selective Pressures from Therapeutics Targeting the Spike Protein
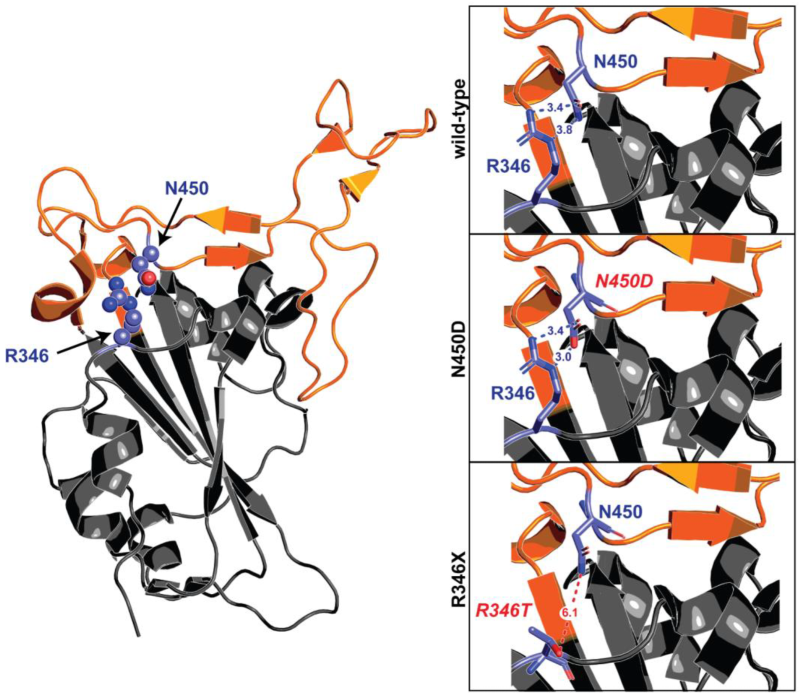
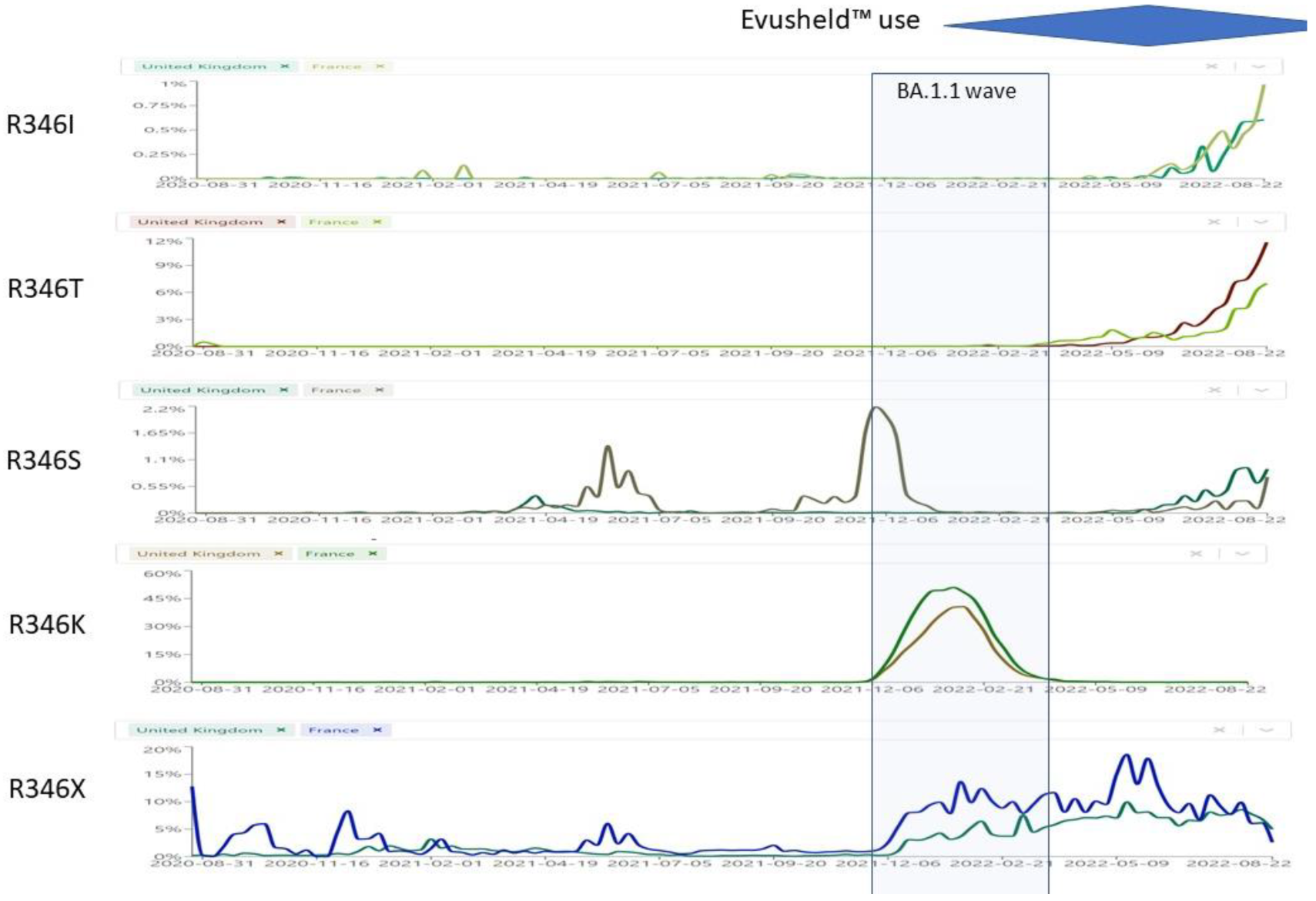
8.1. S:R346X
- R346K (previously seen only in VOC Mu/B.1.621 [48]) occurred exclusively in BA.1.1, a sublineage that disappeared since May 2022, where it affected the interaction network in the BA.1.1 RBD/hACE2 interface through long-range alterations and contributes to the higher hACE2 affinity of the BA.1.1 RBD than the BA.1 RBD [49], and had increased resistance against Evusheld™ [50] and sotrovimab [51]. Only STI-9167 remained effective among the mAbs [52]. Beta + R346K, which was identified in the Philippines in August 2021, exhibited the highest resistance to 2 BNT61b2 doses-elicited sera among the tested VOCs [53]. After BA.1.1, R346K has not been detected worldwide in any sublineage.
- R346I occurs in more than 40 different Omicron sublineages, but it is most represented in BA.5.9 (38%), BA.4.1 (5%), BA.5.9 (4%), but also occurred in AY.39 (14%);
- R346S (previously seen only in a C.36.3 sublineage from Italy [54] (30.8%), occurs in more than 40 different Omicron sublineages but it is most represented in B.1.640.1 (18%), and in a few Delta sublineages (<2%)) occurs nowadays in BA.4.7 (13%), BA.5.2.1 (8.22%), BA.4 (2.8%).
- R346T occurs in more than 96 different Omicron sublineages, but it is mostly represented in BA.4.6 (44%), BA.5.2.1 (13%), BA.2 (8%), BA.2.74 (3%), BA.2.76 (12%), BA.4.1 (2.3%). In addition, it is a hallmark mutation of BA.1.23, BA.2.9.4, BL.1, BA.2.75.2, BA.2.80, BA.2.82, BA.4.1.8, BF.7 and BF.11. BA.4.6, BA.4.7, and BA.5.9 displayed higher humoral immunity evasion capability than BA.4/BA.5, causing 1.5 to 1.9-fold decrease in NT50 of the plasma from BA.1 and BA.2 breakthrough-infection convalescents compared to BA.4/BA.5. Importantly, plasma from BA.5 breakthrough-infection convalescents also exhibits significant neutralization activity decrease against BA.4.6, BA.4.7, and BA.5.9 than BA.4/BA.5, showing on average 2.4 to 2.6-fold decrease in NT50. R346S causes resistance to class 3 antibodies: bebtelovimab remains potent, while Evusheld™ is completely escaped by these subvariants [55].
8.2. S:K444X
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rubin, R. The COVID-19 Pandemic Rages on for People Who Are Immunocompromised. JAMA 2022, 327, 1853–1855. [Google Scholar] [CrossRef] [PubMed]
- Dioverti, V.; Salto-Alejandre, S.; Haidar, G. Immunocompromised Patients with Protracted COVID-19: A Review of “Long Persisters”. Curr. Transplant. Rep. 2022, 9, 209–218. [Google Scholar] [CrossRef]
- Wallace, B.I.; Kenney, B.; Malani, P.N.; Clauw, D.J.; Nallamothu, B.K.; Waljee, A.K. Prevalence of Immunosuppressive Drug Use among Commercially Insured US Adults, 2018–2019. JAMA Netw. Open 2021, 4, e214920. [Google Scholar] [CrossRef]
- Daily New Confirmed COVID-19 Deaths Per Million People. Our World in Data. Available online: https://ourworldindata.org/explorers/coronavirus-data-explorer?zoomToSelection=true&time=2020-03-01..latest&facet=none&pickerSort=asc&pickerMetric=location&hideControls=false&Metric=Confirmed+deaths&Interval=7-day+rolling+average&Relative+to+Population=true&Color+by+test+positivity=false&country=~OWID_WRL (accessed on 1 December 2022).
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Harari, S.; Tahor, M.; Rutsinsky, N.; Meijer, S.; Miller, D.; Henig, O.; Halutz, O.; Levytskyi, K.; Ben-Ami, R.; Adler, A.; et al. Drivers of adaptive evolution during chronic SARS-CoV-2 infections. Nat. Med. 2022, 28, 1501–1508. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.A.J.; Richter, A.; Casey, A.; Osman, H.; Mirza, J.D.; Stockton, J.; Quick, J.; Ratcliffe, L.; Sparks, N.; Cumley, N.; et al. Recurrent SARS-CoV-2 mutations in immunodeficient patients. Virus Evol. 2022, 8, veac050. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D. Molnupiravir: From Hope to Epic Fail? Viruses 2022, 14, 2560. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Lapedes, A.S.; de Jong, J.C.; Bestebroer, T.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Mapping the antigenic and genetic evolution of influenza virus. Science 2004, 305, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Eguia, R.T.; Crawford, K.H.D.; Stevens-Ayers, T.; Kelnhofer-Millevolte, L.; Greninger, A.L.; Englund, J.A.; Boeckh, M.J.; Bloom, J.D. A human coronavirus evolves antigenically to escape antibody immunity. PLoS Pathog. 2021, 17, e1009453. [Google Scholar] [CrossRef]
- Niesen, M.; Anand, P.; Silvert, E.; Suratekar, R.; Pawlowski, C.; Ghosh, P.; Lenehan, P.; Hughes, T.; Zemmour, D.; OHoro, J.C.; et al. COVID-19 vaccines dampen genomic diversity of SARS-CoV-2: Unvaccinated patients exhibit more antigenic mutational variance. medRxiv 2021, 21259833. [Google Scholar] [CrossRef]
- Yeh, T.-Y.; Contreras, G.P. Full vaccination is imperative to suppress SARS-CoV-2 delta variant mutation frequency. medRxiv 2021, 21261768. [Google Scholar] [CrossRef]
- Ruis, C.; Peacock, T.P.; Polo, L.M.; Masone, D.; Alvarez, M.S.; Hinrichs, A.S.; Turakhia, Y.; Cheng, Y.; McBroome, J.; Corbett-Detig, R.; et al. Mutational spectra distinguish SARS-CoV-2 replication niches. bioRxiv 2022, 509649. [Google Scholar] [CrossRef]
- Bloom, J.D.; Beichman, A.C.; Neher, R.A.; Harris, K. Evolution of the SARS-CoV-2 mutational spectrum. bioRxiv 2022, 2022.2011.2019.517207. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Reviews. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maio, N.; Walker, C.R.; Turakhia, Y.; Lanfear, R.; Corbett-Detig, R.; Goldman, N. Mutation Rates and Selection on Synonymous Mutations in SARS-CoV-2. Genome Biol. Evol. 2021, 13, evab087. [Google Scholar] [CrossRef]
- Ratcliff, J.; Simmonds, P. Potential APOBEC-mediated RNA editing of the genomes of SARS-CoV-2 and other coronaviruses and its impact on their longer term evolution. Virology 2021, 556, 62–72. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Takada, K.; Ueda, M.T.; Watanabe, T.; Nakagawa, S. Genomic diversity of SARS-CoV-2 can be accelerated by a mutation in the nsp14 gene. bioRxiv 2020, 424231. [Google Scholar] [CrossRef]
- Jung, C.; Kmiec, D.; Koepke, L.; Zech, F.; Jacob, T.; Sparrer, K.M.J.; Kirchhoff, F. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e02077-21. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D. SARS-CoV-2 Spike Protein Convergent Evolution; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Neto, D.F.L.; Fonseca, V.; Jesus, R.; Dutra, L.H.; Portela, L.M.O.; Freitas, C.; Fillizola, E.; Soares, B.; Abreu, A.L.; Twiari, S.; et al. Molecular dynamics simulations of the SARS-CoV-2 Spike protein and variants of concern: Structural evidence for convergent adaptive evolution. J. Biomol. Struct. Dyn. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, V.; Patrick, C.; Lucas, A.; Mallela, K.M.G. Convergent Evolution of Multiple Mutations Improves the Viral Fitness of SARS-CoV-2 Variants by Balancing Positive and Negative Selection. Biochemistry 2022, 61, 963–980. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200.e7. [Google Scholar] [CrossRef] [PubMed]
- Variant Report 2022-09-14. Available online: https://github.com/neherlab/SARS-CoV-2_variant-reports/blob/main/reports/variant_report_2022-09-14.md (accessed on 23 November 2022).
- Resende, P.C.; Naveca, F.G.; Lins, R.D.; Dezordi, F.Z.; Ferraz, M.V.F.; Moreira, E.G.; Coêlho, D.F.; Motta, F.C.; Paixão, A.C.D.; Appolinario, L.; et al. The ongoing evolution of variants of concern and interest of SARS-CoV-2 in Brazil revealed by convergent indels in the amino (N)-terminal domain of the spike protein. Virus Evol. 2021, 7, veab069. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Zhang, N.; Wang, Y.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2022. [Google Scholar] [CrossRef]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. Bioinformatics 2021, 38, 1735–1737. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [Green Version]
- Focosi, D.; Maggi, F. Do We Really Need Omicron Spike-Based Updated COVID-19 Vaccines? Evidence and Pipeline. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Al Khalaf, R.; Bernasconi, A.; Pinoli, P.; Ceri, S. Analysis of co-occurring and mutually exclusive amino acid changes and detection of convergent and divergent evolution events in SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 4238–4250. [Google Scholar] [CrossRef]
- Kim, I.-J.; Lee, Y.-h.; Khalid, M.M.; Zhang, Y.; Ott, M.; Verdin, E. SARS-CoV-2 ORF8 limits expression levels of Spike antigen. bioRxiv 2022, 515752. [Google Scholar] [CrossRef]
- Akaishi, T.; Fujiwara, K.; Ishii, T. Insertion/deletion hotspots in the Nsp2, Nsp3, S1, and ORF8 genes of SARS-related coronaviruses. BMC Ecol. Evol. 2022, 22, 123. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; McConnell, S.; Casadevall, A.; Cappello, E.; Valdiserra, G.; Tuccori, M. Monoclonal antibody therapies against SARS-CoV-2. Lancet Infect. Dis. 2022, 22, 00311–00315. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Casadevall, A. A Critical Analysis of the Use of Cilgavimab plus Tixagevimab Monoclonal Antibody Cocktail (Evusheld™) for COVID-19 Prophylaxis and Treatment. Viruses 2022, 14, 1999. [Google Scholar]
- Focosi, D.; Maggi, F.; Franchini, M.; McConnell, S.; Casadevall, A. Analysis of Immune Escape Variants from Antibody-Based Therapeutics against COVID-19: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Copin, R.; Baum, A.; Wloga, E.; Pascal, K.E.; Giordano, S.; Fulton, B.O.; Zhou, A.; Negron, N.; Lanza, K.; Chan, N.; et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell 2021, 184, 3949–3961. [Google Scholar] [CrossRef]
- Magnus, C.L.; Hiergeist, A.; Schuster, P.; Rohrhofer, A.; Medenbach, J.; Gessner, A.; Peterhoff, D.; Schmidt, B. Targeted escape of SARS-CoV-2 in vitro from monoclonal antibody S309, the precursor of sotrovimab. Front. Immunol. 2022, 13, 966236. [Google Scholar] [CrossRef]
- Dong, J.; Zost, S.J.; Greaney, A.J.; Starr, T.N.; Dingens, A.S.; Chen, E.C.; Chen, R.E.; Case, J.B.; Sutton, R.E.; Gilchuk, P.; et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat. Microbiol. 2021, 6, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- FDA. Fact Sheet for Healthcare Providers: Emergency Use Authorization for Evusheld™ (Tixagevimab Co-Packaged with Cilgavimab). 2021. Available online: https://www.fda.gov/media/154701/download (accessed on 13 August 2022).
- Lee, J.S.; Yun, K.W.; Jeong, H.; Kim, B.; Kim, M.J.; Park, J.H.; Shin, H.S.; Oh, H.S.; Sung, H.; Song, M.G.; et al. SARS-CoV-2 shedding dynamics and transmission in immunosuppressed patients. Virulence 2022, 13, 1242–1251. [Google Scholar] [CrossRef]
- Andrés, C.; González-Sánchez, A.; Jiménez, M.; Márquez-Algaba, E.; Piñana, M.; Fernández-Naval, C.; Esperalba, J.; Saubi, N.; Quer, J.; Rando-Segura, A.; et al. Emergence of Delta and Omicron variants carrying resistance-associated mutations in immunocompromised patients undergoing Sotrovimab treatment with long viral excretion. Clin. Microbiol. Infect. 2022. [Google Scholar] [CrossRef]
- Gonzalez-Reiche, A.S.; Alshammary, H.; Schaefer, S.; Patel, G.; Polanco, J.; Amoako, A.A.; Rooker, A.; Cognigni, C.; Floda, D.; van de Guchte, A.; et al. Intrahost evolution and forward transmission of a novel SARS-CoV-2 Omicron BA.1 subvariant. medRxiv 2022, 22275533. [Google Scholar] [CrossRef]
- Lee, C.Y.; Shah, M.K.; Hoyos, D.; Solovyov, A.; Douglas, M.; Taur, Y.; Maslak, P.; Babady, N.E.; Greenbaum, B.; Kamboj, M.; et al. Prolonged SARS-CoV-2 Infection in Patients with Lymphoid Malignancies. Cancer Discov. 2022, 12, 62–73. [Google Scholar] [CrossRef]
- Sonnleitner, S.T.; Prelog, M.; Sonnleitner, S.; Hinterbichler, E.; Halbfurter, H.; Kopecky, D.B.C.; Almanzar, G.; Koblmüller, S.; Sturmbauer, C.; Feist, L.; et al. Cumulative SARS-CoV-2 mutations and corresponding changes in immunity in an immunocompromised patient indicate viral evolution within the host. Nat. Commun. 2022, 13, 2560. [Google Scholar] [CrossRef]
- Fratev, F. R346K Mutation in the Mu Variant of SARS-CoV-2 Alters the Interactions with Monoclonal Antibodies from Class 2: A Free Energy Perturbation Study. J. Chem. Inf. Model. 2022, 62, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L.; et al. Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA.2 and BA.1.1. Cell 2022, 185, 2952–2960.e10. [Google Scholar] [CrossRef]
- Uraki, R.; Kiso, M.; Imai, M.; Yamayoshi, S.; Ito, M.; Fujisaki, S.; Takashita, E.; Ujie, M.; Furusawa, Y.; Yasuhara, A.; et al. Therapeutic efficacy of monoclonal antibodies and antivirals against SARS-CoV-2 Omicron BA.1 in Syrian hamsters. Nat. Microbiol. 2022, 7, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent cross-reactive antibodies following Omicron breakthrough in vaccinees. Cell 2022, 185, 2116–2131.e18. [Google Scholar] [CrossRef] [PubMed]
- Duty, J.A.; Kraus, T.; Zhou, H.; Zhang, Y.; Shaabani, N.; Yildiz, S.; Du, N.; Singh, A.; Miorin, L.; Li, D.; et al. Discovery of a SARS-CoV-2 Broadly-Acting Neutralizing Antibody with Activity against Omicron and Omicron + R346K Variants. bioRxiv 2022, 476998. [Google Scholar] [CrossRef]
- Koyama, T.; Miyakawa, K.; Tokumasu, R.; Jeremiah, S.S.; Kudo, M.; Ryo, A. Evasion of vaccine-induced humoral immunity by emerging sub-variants of SARS-CoV-2. Future Microbiol. 2022, 17, 417–424. [Google Scholar] [CrossRef]
- Castelli, M.; Baj, A.; Criscuolo, E.; Ferrarese, R.; Diotti, R.; Sampaolo, M.; Novazzi, F.; Dalla Gasperina, D.; Focosi, D.; Locatelli, M.; et al. Characterization of a lineage C.36 SARS-CoV-2 isolate with reduced susceptibility to neutralization circulating in Lombardy, Italy. Viruses 2021, 13, 1514. [Google Scholar] [CrossRef]
- Jian, F.; Yu, Y.; Song, W.; Yisimayi, A.; Yu, L.; Gao, Y.; Zhang, N.; Wang, Y.; Shao, F.; Hao, X.; et al. Further humoral immunity evasion of emerging SARS-CoV-2 BA.4 and BA.5 subvariants. bioRxiv 2022, 503384. [Google Scholar] [CrossRef] [PubMed]
- Turelli, P.; Fenwick, C.; Raclot, C.; Genet, V.; Pantaleo, G.; Trono, D. P2G3 human monoclonal antibody neutralizes SARS-CoV-2 Omicron subvariants including BA.4 and BA.5 and Bebtelovimab escape mutants. bioRxiv 2022, 501852. [Google Scholar] [CrossRef]
- Umair, M.; Ikram, A.; Salman, M.; Haider, S.A.; Badar, N.; Rehman, Z.; Ammar, M.; Rana, M.S.; Ali, Q. Genomic surveillance reveals the detection of SARS-CoV-2 delta, beta, and gamma VOCs during the third wave in Pakistan. J. Med. Virol. 2022, 94, 1115–1129. [Google Scholar] [CrossRef]
- Ortega, J.T.; Pujol, F.H.; Jastrzebska, B.; Rangel, H.R. Mutations in the SARS-CoV-2 spike protein modulate the virus affinity to the human ACE2 receptor, an in silico analysis. EXCLI J. 2021, 20, 585–600. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 28, e61312. [Google Scholar] [CrossRef] [PubMed]
- Saifi, S.; Ravi, V.; Sharma, S.; Swaminathan, A.; Chauhan, N.S.; Pandey, R. SARS-CoV-2 VOCs, Mutational diversity and clinical outcome: Are they modulating drug efficacy by altered binding strength? Genomics 2022, 114, 110466. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef]
- Roemer, C.H.; Hisner, R.; Frohberg, N.; Sakaguchi, H.; Gueli, F.; Peacock, T. SARS-CoV-2 Evolution, Post-Omicron. Available online: https://virological.org/t/sars-cov-2-evolution-post-omicron/911 (accessed on 26 November 2022).
- Lambisia, A.; Nyiro, J.; Morobe, J.; Makori, T.; Ndwiga, L.; Mburu, M.; Moraa, E.; Musyoki, J.; Murunga, N.; Bejon, P.; et al. Detection of a SARS-CoV-2 Beta-like Variant with Additional Mutations in Coastal Kenya after >1 Year of Disappearance. Available online: https://virological.org/t/detection-of-a-sars-cov-2-beta-like-variant-with-additional-mutations-in-coastal-kenya-after-1-year-of-disappearance/910 (accessed on 26 November 2022).
- Sullivan, D.J.; Franchini, M.; Joyner, M.J.; Casadevall, A.; Focosi, D. Analysis of anti-Omicron neutralizing antibody titers in different convalescent plasma sources. Nat. Commun. 2022, 13, 6478. [Google Scholar] [CrossRef]
- Sullivan, D.J.; Franchini, M.; Senefeld, J.W.; Joyner, M.J.; Casadevall, A.; Focosi, D. Plasma after both SARS-CoV-2 boosted vaccination and COVID-19 potently neutralizes BQ1.1 and XBB. bioRxiv 2022, 517977. [Google Scholar] [CrossRef]
- FDA Announces Bebtelovimab is Not Currently Authorized in Any US Region. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-announces-bebtelovimab-not-currently-authorized-any-us-region (accessed on 1 December 2022).
- FDA Statement. January 24, 2022. Coronavirus (COVID-19) Update: FDA Limits Use of Certain Monoclonal Antibodies to Treat COVID-19 Due to the Omicron Variant. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-limits-use-certain-monoclonal-antibodies-treat-covid-19-due-omicron (accessed on 3 February 2022).
- FDA Updates Sotrovimab Emergency Use Authorization. March 30, 2022. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-updates-sotrovimab-emergency-use-authorization (accessed on 26 April 2022).
- FDA Releases Important Information about Risk of COVID-19 Due to Certain Variants Not Neutralized by Evusheld. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-releases-important-information-about-risk-covid-19-due-certain-variants-not-neutralized-evusheld (accessed on 10 October 2022).
- Senefeld, J.W.; Klassen, S.A.; Ford, S.K.; Senese, K.A.; Wiggins, C.C.; Bostrom, B.C.; Thompson, M.A.; Baker, S.E.; Nicholson, W.T.; Johnson, P.W.; et al. Use of convalescent plasma in COVID-19 patients with immunosuppression. Transfusion 2021, 61, 2503–2511. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spike Mutation | Main Lineages | BAM | ETE | CAS | IMD | CIL | TIX | SOT | BEB | REG | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| R346X | T | BA.2.3.22, BS.1.*, BP.1, DD.1, BJ.1, BL.1.*, BL.2.*, BL.5, BA.2.75.2.* (CA.*), BM.1.1.* (CJ.* and CV.*), BM.4.1.1.1.* (CH.*), BR.2.* and BR.3, BN.1, BA.2.75.6.* (BY.*), BA.2.75.9.* (CY.*), BA.2.76, BA.4.1.8 and BA.4.1.9, CS.1, BA.4.6.* (DC.*) and BA.4.7, BA.5.1.18 and BA.5.1.20, DE.2, BA.5.1.26.* (CU.*), BA.5.1.27 and BA.5.1.28, BF.7.*, BF.11.*, BA.5.2.6.* (CP.*), BA.5.2.13.* (CR.*), BA.5.2.25.* (DA.*), BA.5.2.39, BQ.1.1.* (CZ.*, CW.*, DK.*), BE.1.2.*, BE.1.4.2, BE.4.1.* (CQ.*), BE.5, BE.6, BE.7, BF.1, CK.1.2, CM.11, BL.6, XBB.*, XBD, XBE, XBF, XBG | |||||||||
| E | BA.5.6.4 | ||||||||||
| I | BF.33, CE.1 | ||||||||||
| K | BA.1.1 | ||||||||||
| R | BA.5.2.25, DB.2 | ||||||||||
| S | BL.5, BF.13, BQ.1.21, BE.6 | ||||||||||
| K444X | M | CA.3.1, BR.1.*, BA.5.2.7, CY.1, BU.1, CG.1, BQ.1.17 | |||||||||
| N | BA.2.38.*, BA.4.6.3, BA.5.1.29, BV.2, BA.5.2.24, CK.* (DG.*), BE.4.2 | ||||||||||
| R | BA.2.3.20.* (CM.*), CS.1, BF.16, BA.5.2.18, CR.1.*, CR.2, BA.5.2.41, CQ.1.*, XBB.4.* | ||||||||||
| T | CH.1.*, BR.4, BA.5.2.25, DB.1, DB.2, BA.5.2.36.* (CT.1), BE.1.1.1, BQ.1.* (CZ.*, CW.*, DK.*), BQ.2, BE.9, BA.5.2.46, BA.5.6.2.* (BW.1) | ||||||||||
| V445 | A | BA.4.6.2, BF.25, CP.1.1, BU.2, CR.1.2, BA.5.2.23, BE.1.2.1, BE.1.4.3, CQ.2 | |||||||||
| P | BJ.1, XBB.* | ||||||||||
| G446 | D | BA.5.2.30, CD.1 | |||||||||
| G | BR.4 | ||||||||||
| S | BA.1.*, CM.8.*, BJ.1, BA.2.10.4, BH.1, BA.2.75.* (BL.*, CA.*, BM.*, CJ.*, CV.*, CH.*, BR.*, BN.*, BY.*, CB.*), BF.3.1, CP.1.3, CQ.1, XBB.*, XBC, XBD, XBF | ||||||||||
| N450D | BU.3, CN.1, BA.5.2.32, BA.5.2.40, CC.1 | ||||||||||
| L452X | L | XBD | |||||||||
| M | BP.1, BA.2.3.20.* (CM.*), XBC.1 | ||||||||||
| Q | BH.1, BA.2.75.8 | ||||||||||
| R | BS.1.*, CA.1, CA.3.1, CA.7, CV.1, CH.1.1, BA.2.75.4.* (BR.*), BY.1.1.*, BA.4.* (CS.*, DC.*), BA.5.* (BT.*, DH.*, DE.*, CU.*, CL.*, BF.*, BZ.*, CP.*, CY.*, BU.*, CR.*, BV.*, CN.*, CK.*, DG.*, DB.*, CG.*, CF.*, CD.*, CE.*, CT.*, DA.*, BE.*, BQ.*, CZ.*, CW.*, CC.*, CQ.*, BW.*, DK.*), XBE, XBG | ||||||||||
| N460X | K | BS.1.*, BA.2.3.20.* (CM.*), DD.1, BA.2.75.* (BL.*, CA.*, BM.*, CJ.*, CV.*, CH.*, BR.*, BN.*, BY.*, CB.*), BA.4.6.3, CL.1, BF.33, CY.1, BU.1, CK.1, CK.2.*, DG.1, CK.3, DB.1, BQ.1.* (CZ.*, CW.*, DK.*), BE.4.2, BE.9, BW.1, BA.5.2.46, XBB.*, XBD, XBF | |||||||||
| S | DC.1 | ||||||||||
| Y | CP.3 | ||||||||||
| F486X | I | BM.2.3, BR.2.*, BF.7.12, BF.12 | |||||||||
| P | BA.2.10.4, CA.4, CJ.1, XBB.1.5, XBB.1.9.1, XBB.6.1, XBC.*, XBF, XBL | ||||||||||
| S | BA.2.75.2.* (CA.*), BM.1.* (CV.*), BM.4.1.* (CH.*), BR.1.2, BY.1.*, BA.2.75.7, CM.11, DS.1, XBB.*, XBD | ||||||||||
| V | BM.2.1, CB.1, BA.4.* (CS.*, DC.*), BA.5.* (BT.*, DH.*, DE.*, CU.*, CL.*, BF.*, BZ.*, CP.*, CY.*, BU.*, CR.*, BV.*, CN.*, CK.*, DG.*, DB.*, CG.*, CF.*, CD.*, CE.*, CT.*, DA.*, BE.*, BQ.*, CZ.*, CW.*, CC.*, CQ.*, BW.*, DK.*), XBE, XBG | ||||||||||
| F490X | I | CZ.1 | |||||||||
| L | BL.1.3 | ||||||||||
| S | BM.1.1.1.* (CJ.1), BN.1.*, BN.2.1., BN.3.1, BN.4, XBB.*, XBF | ||||||||||
| V | BJ.1, BL.1.4 | ||||||||||
| R493X | L | BA.2.3.21.1 | |||||||||
| Q | BA.2.10.4, BA.2.75.* (BL.*, CA.*, BM.*, CJ.*, CV.*, CH.*, BR.*, BN.*, BY.*, CB.*), BA.4.* (CS.*, DC.*), BA.5.* (BT.*, DH.*, DE.*, CU.*, CL.*, BF.*, BZ.*, CP.*, CY.*, BU.*, CR.*, BV.*, CN.*, CK.*, DG.*, DB.*, CG.*, CF.*, CD.*, CE.*, CT.*, DA.*, BE.*, BQ.*, CZ.*, CW.*, CC.*, CQ.*, BW.*, DK.*), XBB.*, XBC.*, XBD, XBE, XBF, XBG | ||||||||||
| S494P | BA.2.10.4, CA.2, BN.1.*, BY.1.2.1, BQ.1.1.11, BQ.1.1.12, BQ.1.19 | ||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Focosi, D.; Quiroga, R.; McConnell, S.; Johnson, M.C.; Casadevall, A. Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. Int. J. Mol. Sci. 2023, 24, 2264. https://doi.org/10.3390/ijms24032264
Focosi D, Quiroga R, McConnell S, Johnson MC, Casadevall A. Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. International Journal of Molecular Sciences. 2023; 24(3):2264. https://doi.org/10.3390/ijms24032264
Chicago/Turabian StyleFocosi, Daniele, Rodrigo Quiroga, Scott McConnell, Marc C. Johnson, and Arturo Casadevall. 2023. "Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge" International Journal of Molecular Sciences 24, no. 3: 2264. https://doi.org/10.3390/ijms24032264