



The Role of Arginine-Vasopressin in Stroke and the Potential Use of Arginine-Vasopressin Type 1 Receptor Antagonists in Stroke Therapy: A Narrative Review
 , ,
, ,
Abstract
:1. Introduction
2. The Physiological Role of Arginine Vasopressin
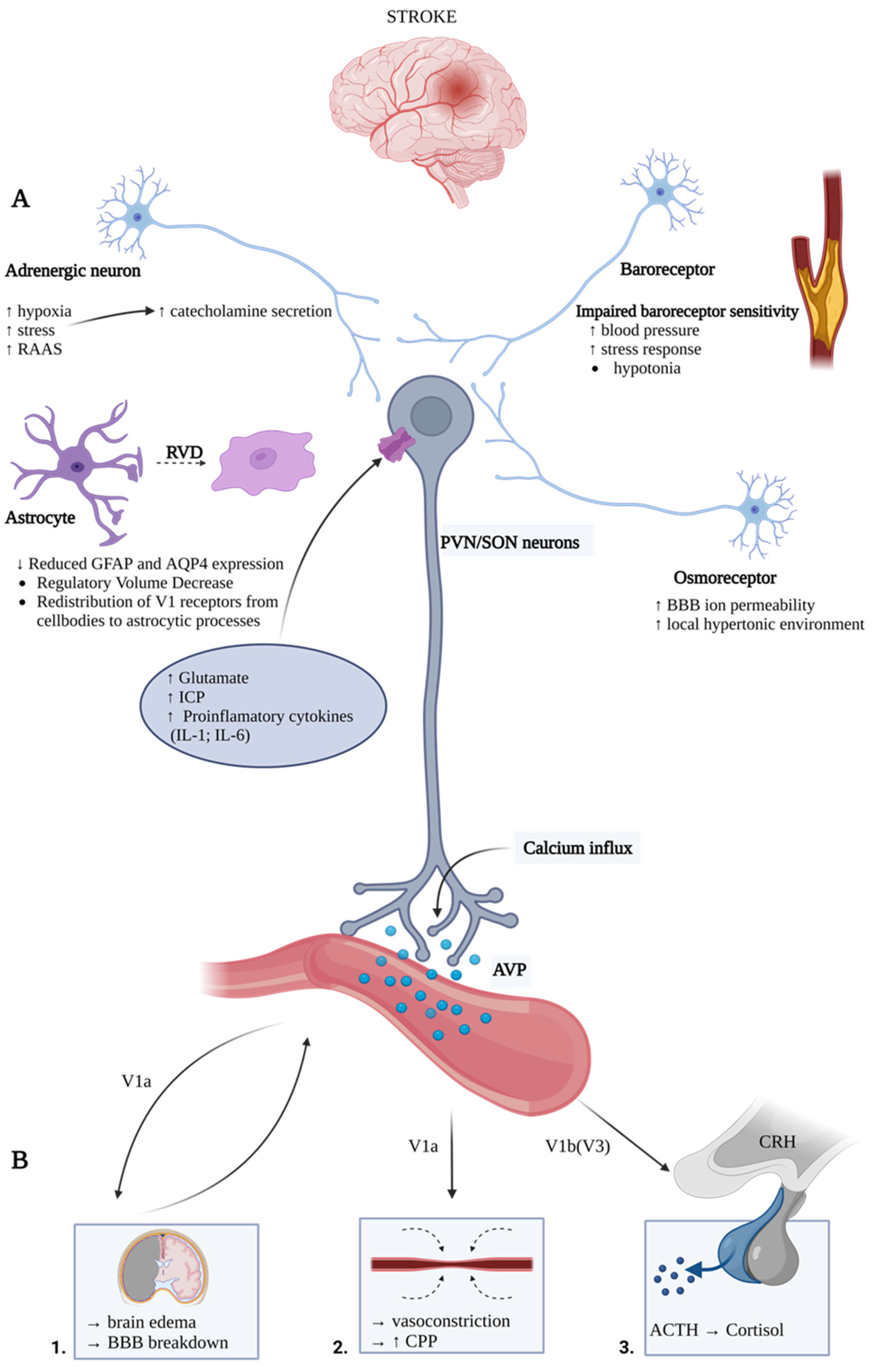
3. Arginine Vasopressin Hypersecretion and Release Mechanisms during Stroke
4. Arginine Vasopressin Contribution to Ischemic Stroke Pathomechanism
4.1. Systemic Effects of AVP in Stroke
4.1.1. Arginine Vasopressin and Hyponatremia
4.1.2. Arginine Vasopressin, Autonomic Nervous System, Inflammation, and Immunosuppression Response
4.1.3. Arginine Vasopressin and Stress Response
4.1.4. Arginine Vasopressin and Platelet Aggregation
4.2. Central Nervous System Local Effects of Arginine Vasopressin System in Stroke

{kind=link}
| Mechanism | Involved Receptors | Receptor Location in the Brain | Effects | Authors |
|---|---|---|---|---|
| Brain Edema Formation | V1aR | Neurons, Glia |
| [6,33,133,153,154,155,156] |
| Time-dependent Arteries and arterioles vasodilation and vasoconstriction. | V1aR | Smooth muscle layer of brain vasculature; Endothelium |
| [145,146,157,158,159,160] |
| Increased BBB permeability | V1aR | Astrocytes, podocytes, brain endothelium |
| [144,145,156,161,162] |
| AVP potentiating action on the CRH-evoked ACTH secretion. | V1bR (V3) | Adenohypophysis |
| [163,164] |
| Post-stroke, edema-associated inflammation | V1aR | Astrocytes |
| [92,150,165] |
5. The Prognostic Value of Copeptin in Acute Stroke Patients
6. Targeting Arginine Vasopressin Receptors in the Acute Phase of Ischemic Stroke
7. Discussion
| Experimental Model/Type of Study | Antagonist (Dose) | Time of Administration | Results | Author |
|---|---|---|---|---|
| Mouse; 60 min tMCAO | Conivaptan (i.v 0.2 mg, 0.2 mg) and Tolvaptan (p.o 0.2 mg) | At reperfusion |
| [179] |
| Mouse; 60 min tMCAO | V1R antagonist (i. cv. 500 ng, 1000 ng) V2R antagonist (i. cv. 1000 ng) | 10 min after surgery |
| [134] |
| Rat; 2 h tMCAO | SR49059 (6480, 720, 640 µL/h) | 1 h before/after reperfusion |
| [29] |
| Mouse; ICH; collagenase injection model | SR49059 (0.5 mg/kg, 2 mg/kg) | 1 h after surgery |
| [135] |
| Mouse; 60 min MCAO | V1R antagonist (i.cv)V2R antagonist (i.cv) | 5 min after MCAO |
| [180] |
| Rat; pMCAO | SR49059 (i.p. 30 mg/kg, 2 mg/kg) | Immediately, 1 h, 3 h, 6 h after MCAO |
| [30] |
| Rat; general cerebral hypoxia; bilateral common carotid ligation model | OPC-31260 (p.o 30 mg/kg) | Immediately after operation |
| [28] |
| Rat; SAH; autologous blood administration on cerebral cortex | OPC-31260 (p.o 30 mg/kg, 10 mg/kg) | Immediately after operation or every 8 h following operation |
| [27] |
| Rat; MCAO | 5% HS bolus +5% HS (HS) Conivaptan (Con) Conivaptan + 5% HS (Con+HS) Conivaptan + 5% HS bolus+ 5% HS (Con+HSb) | 6 h after MCAO—early group 24 h after MCAO—late group |
| [186] |
| Rat; 30 min tCCAO | Conivaptan (i.v. 10 mg/mL, 20 mg/mL) | At reperfusion |
| [183] |
| Mouse; 60 min tMCAO | Conivaptan (i.v. 0.2 mg/day) | 3, 5, and 20 h after reperfusion |
| [182] |
| Phase I clinical study, ICH at risk of developing PHE | Conivaptan (20 mg) + standard management | Every 12 h for 2 days |
| [184] |
| Case report; refractory brain edema following hemorrhagic stroke | Conivaptan (i.v 20 mg in 30 min + 20 mg over 24 h) | 2 days after hemorrhage detection |
| [185] |
8. Conclusions
- A stroke is associated with the release of AVP into the blood in response to various endogenous stimuli;
- Overproduction of AVP is most likely detrimental to the brain, primarily due to the AVP-elicited brain edema;
- Copeptin is a surrogate marker of AVP and can be used as a prognostic biomarker of stroke outcome;
- AVP receptor antagonists can potentially be used as an add-on therapeutic intervention in stroke.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global, regional, and national burden of stroke and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet. Neurol. 2021, 20, 795–820. [CrossRef] [PubMed]
- Matei, N.; Camara, J.; Zhang, J.H. The Next Step in the Treatment of Stroke. Front. Neurol. 2021, 11, 582605. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, S.M.; Ong, L.K.; Collins-Praino, L.E.; Turner, R.J. Neuroinflammation as a Key Driver of Secondary Neurodegeneration Following Stroke? Int. J. Mol. Sci. 2021, 22, 13101. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Berl, T.; Anderson, R.J. Osmotic and nonosmotic control of vasopressin release. Am. J. Physiol. 1979, 236, F321–F332. [Google Scholar] [CrossRef]
- Kozniewska, E.; Romaniuk, K. Vasopressin in vascular regulation and water homeostasis in the brain. J. Physiol. Pharmacol. Off. J. Polish Physiol. Soc. 2008, 59 (Suppl 8), 109–116. [Google Scholar]
- Szmydynger-Chodobska, J.; Chung, I.; Koźniewska, E.; Tran, B.; Harrington, F.; Duncan, J.; Chodobski, A. Increased Expression of Vasopressin V 1a Receptors after Traumatic Brain Injury. J. Neurotrauma 2004, 21, 1090–1102. [Google Scholar] [CrossRef]
- Sakka, L.; Coll, G.; Chazal, J. Anatomy and physiology of cerebrospinal fluid. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2011, 128, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Bankir, L.; Bichet, D.G.; Morgenthaler, N.G. Vasopressin: Physiology, assessment and osmosensation. J. Intern. Med. 2017, 282, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Danziger, J.; Zeidel, M.L. Osmotic Homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 852–862. [Google Scholar] [CrossRef] [Green Version]
- Toba, K.; Ohta, M.; Kimura, T.; Nagano, K.; Ito, S.; Ouchi, Y. Role of brain vasopressin in regulation of blood pressure. In Advances in Brain Vasopressin; Urban, I.J.A., Burbach, J.P.H., De Wed, D., Eds.; Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 1999; Volume 119, pp. 337–349. [Google Scholar]
- Jia, S.W.; Liu, X.Y.; Wang, S.C.; Wang, Y.F. Vasopressin hypersecretion-associated brain edema formation in ischemic stroke: Underlying mechanisms. J. Stroke Cerebrovasc. Dis. 2016, 25, 1289–1300. [Google Scholar] [CrossRef]
- Burton, T.M.; Bushnell, C.D. Reversible Cerebral Vasoconstriction Syndrome. Stroke 2019, 50, 2253–2258. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke Off. J. Int. Stroke Soc. 2009, 4, 461–470. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Martino, M.; Arnaldi, G. Copeptin and Stress. Endocrines 2021, 2, 384–404. [Google Scholar] [CrossRef]
- Acher, R.; Chauvet, J.; Rouille, Y. Dynamic processing of neuropeptides: Sequential conformation shaping of neurohypophysial preprohormones during intraneuronal secretory transport. J. Mol. Neurosci. 2002, 18, 223–228. [Google Scholar] [CrossRef]
- Morgenthaler, N.G.; Struck, J.; Alonso, C.; Bergmann, A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin. Chem. 2006, 52, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Christ-Crain, M. Vasopressin and Copeptin in health and disease. Rev. Endocr. Metab. Disord. 2019, 20, 283–294. [Google Scholar] [CrossRef]
- Wendt, M.; Ebinger, M.; Kunz, A.; Rozanski, M.; Waldschmidt, C.; Weber, J.E.; Winter, B.; Koch, P.M.; Nolte, C.H.; Hertel, S.; et al. Copeptin Levels in Patients With Acute Ischemic Stroke and Stroke Mimics. Stroke 2015, 46, 2426–2431. [Google Scholar] [CrossRef] [Green Version]
- De Marchis, G.M.; Katan, M.; Weck, A.; Fluri, F.; Foerch, C.; Findling, O.; Schuetz, P.; Buhl, D.; El-Koussy, M.; Gensicke, H.; et al. Copeptin adds prognostic information after ischemic stroke: Results from the CoRisk study. Neurology 2013, 80, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- De Marchis, G.M.; Weck, A.; Audebert, H.; Benik, S.; Foerch, C.; Buhl, D.; Schuetz, P.; Jung, S.; Seiler, M.; Morgenthaler, N.G.; et al. Copeptin for the Prediction of Recurrent Cerebrovascular Events After Transient Ischemic Attack. Stroke 2014, 45, 2918–2923. [Google Scholar] [CrossRef] [Green Version]
- Perovic, E.; Mrdjen, A.; Harapin, M.; Kuna, A.T.; Simundic, A.-M. Diagnostic and prognostic role of resistin and copeptin in acute ischemic stroke. Top. Stroke Rehabil. 2017, 24, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Purroy, F.; Suárez-Luis, I.; Cambray, S.; Farré, J.; Benabdelhak, I.; Mauri-Capdevila, G.; Sanahuja, J.; Quílez, A.; Begué, R.; Gil, M.I.; et al. The determination of copeptin levels helps management decisions among transient ischaemic attack patients. Acta Neurol. Scand. 2016, 134, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tian, Y.; Peng, H.; Li, H. Copeptin as a biomarker for prediction of prognosis of acute ischemic stroke and transient ischemic attack: A meta-analysis. Hypertens. Res. 2017, 40, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Greisenegger, S.; Segal, H.C.; Burgess, A.I.; Poole, D.L.; Mehta, Z.; Rothwell, P.M. Copeptin and Long-Term Risk of Recurrent Vascular Events After Transient Ischemic Attack and Ischemic Stroke. Stroke 2015, 46, 3117–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, L.; Xu, J.; Chen, Y.; Gibbs, M.E.; Du, T.; Hertz, L.; Xu, J.; Chen, Y.; Gibbs, M.E.; Du, T. Antagonists of the Vasopressin V1 Receptor and of the β(1)-Adrenoceptor Inhibit Cytotoxic Brain Edema in Stroke by Effects on Astrocytes-but the Mechanisms Differ. Curr. Neuropharmacol. 2014, 12, 308–323. [Google Scholar] [CrossRef] [Green Version]
- László, F.A.; Varga, C.; Nakamura, S. Vasopressin receptor antagonist OPC-31260 prevents cerebral oedema after subarachnoid haemorrhage. Eur. J. Pharmacol. 1999, 364, 115–122. [Google Scholar] [CrossRef]
- Molnár, A.H.; Varga, C.; Berkó, A.; Rojik, I.; Párducz, A.; László, F.; László, F.A. Inhibitory effect of vasopressin receptor antagonist OPC-31260 on experimental brain oedema induced by global cerebral ischaemia. Acta Neurochir. 2008, 150, 265–271. [Google Scholar] [CrossRef]
- Kleindienst, A.; Fazzina, G.; Dunbar, J.G.; Glisson, R.; Marmarou, A. Protective effect of the V1a receptor antagonist SR49059 on brain edema formation following middle cerebral artery occlusion in the rat. Acta Neurochir. Suppl. 2006, 4, 303–306. [Google Scholar] [CrossRef]
- Shuaib, A.; Wang, C.X.; Yang, T.; Noor, R. Effects of nonpeptide V1 vasopressin receptor antagonist SR-49059 on infarction volume and recovery of function in a focal embolic stroke model. Stroke 2002, 33, 3033–3037. [Google Scholar] [CrossRef] [Green Version]
- Marmarou, C.R.; Liang, X.; Abidi, N.H.; Parveen, S.; Taya, K.; Henderson, S.C.; Young, H.F.; Filippidis, A.S.; Baumgarten, C.M. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014, 1581, 89–102. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, S.; Amiry-Moghaddam, M.; Ottersen, O.P.; Bhardwaj, A. Conivaptan, a Selective Arginine Vasopressin V1a and V2 Receptor Antagonist Attenuates Global Cerebral Edema Following Experimental Cardiac Arrest via Perivascular Pool of Aquaporin-4. Neurocrit. Care 2016, 24, 273–282. [Google Scholar] [CrossRef]
- Okuno, K.; Taya, K.; Marmarou, C.R.; Ozisik, P.; Fazzina, G.; Kleindienst, A.; Gulsen, S.; Marmarou, A. The modulation of aquaporin-4 by using PKC-activator (phorbol myristate acetate) and V1a receptor antagonist (SR49059) following middle cerebral artery occlusion/reperfusion in the rat. In Proceedings of the Acta Neurochirurgica Supplements; Steiger, H.-J., Ed.; Springer: Vienna, Austria, 2009; pp. 431–436. [Google Scholar]
- Rajan, S.; Tosh, P.; Kadapamannil, D.; Srikumar, S.; Paul, J.; Kumar, L. Efficacy of vaptans for correction of postoperative hyponatremia: A comparison between single intravenous bolus conivaptan vs oral tolvaptan. J. Anaesthesiol. Clin. Pharmacol. 2018, 34, 193–197. [Google Scholar] [CrossRef]
- Ameli, P.A.; Ameli, N.J.; Gubernick, D.M.; Ansari, S.; Mohan, S.; Satriotomo, I.; Buckley, A.K.; Maxwell, C.W.; Nayak, V.H.; Shushrutha Hedna, V. Role of vasopressin and its antagonism in stroke related edema. J. Neurosci. Res. 2014, 92, 1091–1099. [Google Scholar] [CrossRef]
- Juul, K.V.; Bichet, D.G.; Nielsen, S.; Nørgaard, J.P. The physiological and pathophysiological functions of renal and extrarenal vasopressin V2 receptors. Am. J. Physiol. Renal Physiol. 2014, 306, F931–F940. [Google Scholar] [CrossRef]
- García-Villalón, A.L.; Garcia, J.L.; Fernández, N.; Monge, L.; Gómez, B.; Diéguez, G. Regional differences in the arterial response to vasopressin: Role of endothelial nitric oxide. Br. J. Pharmacol. 1996, 118, 1848–1854. [Google Scholar] [CrossRef]
- Aoyagi, T.; Koshimizu, T.A.; Tanoue, A. Vasopressin regulation of blood pressure and volume: Findings from V1a receptor-deficient mice. Kidney Int. 2009, 76, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Yamamura, Y.; Chihara, T.; Onogawa, T.; Nakamura, S.; Yamashita, T.; Mori, T.; Tominaga, M.; Yabuuchi, Y. OPC-21268, a vasopressin V1 antagonist, produces hypotension in spontaneously hypertensive rats. Hypertension 1994, 23, 200–204. [Google Scholar] [CrossRef] [Green Version]
- Friesenecker, B.E.; Tsai, A.G.; Martini, J.; Ulmer, H.; Wenzel, V.; Hasibeder, W.R.; Intaglietta, M.; Dünser, M.W. Arteriolar vasoconstrictive response: Comparing the effects of arginine vasopressin and norepinephrine. Crit. Care 2006, 10, R75. [Google Scholar] [CrossRef] [Green Version]
- Sedhai, Y.R.; Shrestha, D.B.; Budhathoki, P.; Memon, W.; Acharya, R.; Gaire, S.; Pokharel, N.; Maharjan, S.; Jasaraj, R.; Sodhi, A.; et al. Vasopressin versus norepinephrine as the first-line vasopressor in septic shock: A systematic review and meta-analysis. J. Clin. Transl. Res. 2022, 8, 185–199. [Google Scholar]
- Fassbender, K.; Schmidt, R.; Mössner, R.; Daffertshofer, M.; Hennerici, M. Pattern of activation of the hypothalamic-pituitary-adrenal axis in acute stroke. Relation to acute confusional state, extent of brain damage, and clinical outcome. Stroke 1994, 25, 1105–1108. [Google Scholar] [CrossRef] [Green Version]
- Zelena, D.; Pintér, O.; Balázsfi, D.G.; Langnaese, K.; Richter, K.; Landgraf, R.; Makara, G.B.; Engelmann, M. Vasopressin signaling at brain level controls stress hormone release: The vasopressin-deficient Brattleboro rat as a model. Amino Acids 2015, 47, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Thibonnier, M.; Preston, J.A.; Dulin, N.; Wilkins, P.L.; Berti-Mattera, L.N.; Mattera, R. The human V3 pituitary vasopressin receptor: Ligand binding profile and density-dependent signaling pathways. Endocrinology 1997, 138, 4109–4122. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Thompson, C.J. Chapter 18—Vasopressin, Diabetes Insipidus, and the Syndrome of Inappropriate Antidiuresis. In Endocrinology: Adult and Pediatric, 7th ed.; Jameson, J.L., De Groot, L.J., de Kretser, D.M., Giudice, L.C., Grossman, A.B., Melmed, S., Potts, J.T., Weir, G.C., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2016; pp. 298–311.e4. ISBN 978-0-323-18907-1. [Google Scholar]
- Perraudin, V.; Delarue, C.; Lefebvre, H.; Contesse, V.; Kuhn, J.M.; Vaudry, H. Vasopressin stimulates cortisol secretion from human adrenocortical tissue through activation of V1 receptors. J. Clin. Endocrinol. Metab. 1993, 76, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Niermann, H.; Amiry-Moghaddam, M.; Holthoff, K.; Witte, O.W.; Ottersen, O.P. A Novel Role of Vasopressin in the Brain: Modulation of Activity-Dependent Water Flux in the Neocortex. J. Neurosci. 2001, 21, 3045–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabarean, I. V Activation of Preoptic Arginine Vasopressin Neurons Induces Hyperthermia in Male Mice. Endocrinology 2020, 162, bqaa217. [Google Scholar] [CrossRef] [PubMed]
- Buijs, R.M.; De Vries, G.J.; Van Leeuwen, F.W.; Swaab, D.F. Vasopressin and Oxytocin: Distribution and Putative Functions in the Brain. In The Neurohypophysis: Structure, Function and Control; Cross, B.A., Leng, G., Eds.; Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 1983; Volume 60, pp. 115–122. [Google Scholar]
- Kasting, N.W.; Veale, W.L.; Cooper, K.E. Convulsive and hypothermic effects of vasopressin in the brain of the rat. Can. J. Physiol. Pharmacol. 1980, 58, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; van Heerikhuize, J.J.; Wortel, J.; Buijs, R.M. A Diurnal Rhythm of Stimulatory Input to the Hypothalamo{\textendash}Pituitary{\textendash}Adrenal System as Revealed by Timed Intrahypothalamic Administration of the Vasopressin V1Antagonist. J. Neurosci. 1996, 16, 5555–5565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalsbeek, A.; Fliers, E.; Hofman, M.A.; Swaab, D.F.; Buijs, R.M. Vasopressin and the output of the hypothalamic biological clock. J. Neuroendocrinol. 2010, 22, 362–372. [Google Scholar] [CrossRef]
- Gomez, F.; Chapleur, M.; Fernette, B.; Burlet, C.; Nicolas, J.P.; Burlet, A. Arginine vasopressin (AVP) depletion in neurons of the suprachiasmatic nuclei affects the AVP content of the paraventricular neurons and stimulates adrenocorticotrophic hormone release. J. Neurosci. Res. 1997, 50, 565–574. [Google Scholar] [CrossRef]
- Rohr, K.E.; Telega, A.; Savaglio, A.; Evans, J.A. Vasopressin regulates daily rhythms and circadian clock circuits in a manner influenced by sex. Horm. Behav. 2021, 127, 104888. [Google Scholar] [CrossRef]
- Marcinkowska, A.B.; Biancardi, V.C.; Winklewski, P.J. Arginine-vasopressin, synaptic plasticity and brain networks. Curr. Neuropharmacol. 2022, 20, 2292–2302. [Google Scholar] [CrossRef]
- Bosch, O.J.; Neumann, I.D. Brain vasopressin is an important regulator of maternal behavior independent of dams’ trait anxiety. Proc. Natl. Acad. Sci. USA 2008, 105, 17139–17144. [Google Scholar] [CrossRef]
- Neumann, I.D.; Landgraf, R. Balance of brain oxytocin and vasopressin: Implications for anxiety, depression, and social behaviors. Trends Neurosci. 2012, 35, 649–659. [Google Scholar] [CrossRef]
- Boer, G.J.; Buijs, R.M.; Swaab, D.F.; De Vries, G.J. Vasopressin and the developing rat brain. Peptides 1980, 1, 203–209. [Google Scholar] [CrossRef]
- Katan, M.; Christ-Crain, M. The stress hormone copeptin: A new prognostic biomarker in acute illness. Swiss Med. Wkly. 2010, 140, w13101. [Google Scholar] [CrossRef]
- Morgenthaler, N.G.; Müller, B.; Struck, J.; Bergmann, A.; Redl, H.; Christ-Crain, M. Copeptin, a stable peptide of the arginine vasopressin precursor, is elevated in hemorrhagic and septic shock. Shock 2007, 28, 219–226. [Google Scholar] [CrossRef]
- Liu, X.; Jin, Y.; Zheng, H.; Chen, G.; Tan, B.; Wu, B. Arginine vasopressin gene expression in supraoptic nucleus and paraventricular nucleus of hypothalamous following cerebral ischemia and reperfusion. Chinese Med. Sci. J. 2000, 15, 157–161. [Google Scholar]
- Szmydynger-Chodobska, J.; Zink, B.J.; Chodobski, A. Multiple sites of vasopressin synthesis in the injured brain. J. Cereb. blood flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2011, 31, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Barreca, T.; Gandolfo, C.; Corsini, G.; Del Sette, M.; Cataldi, A.; Rolandi, E.; Franceschini, R. Evaluation of the secretory pattern of plasma arginine vasopressin in stroke patients. Cerebrovasc. Dis. 2001, 11, 113–118. [Google Scholar] [CrossRef]
- Loesch, A. The effect of cerebral ischemia on the ultrastructure of the hypothalamo-neurohypophysial system of the mongolian gerbil. The neurohypophysial axons and pituicytes. Aust. J. Exp. Biol. Med. Sci. 1983, 61, 557–568. [Google Scholar] [CrossRef]
- Hockel, K.; Schöller, K.; Trabold, R.; Nussberger, J.; Plesnila, N. Vasopressin V1a Receptors Mediate Posthemorrhagic Systemic Hypertension Thereby Determining Rebleeding Rate and Outcome After Experimental Subarachnoid Hemorrhage. Stroke 2012, 43, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Chen, T.Y.; Chen, C.H.; Crain, B.J.; Toung, T.J.K.; Bhardwaj, A. Plasma arginine-vasopressin following experimental stroke: Effect of osmotherapy. J. Appl. Physiol. 2006, 100, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroja, M.M.C.; Reid, E.; McCabe, C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp. Transl. Stroke Med. 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Johanson, C.E.; Preston, J.E.; Chodobski, A.; Stopa, E.G.; Szmydynger-Chodobska, J.; McMillan, P.N. AVP V1 receptor-mediated decrease in Cl- efflux and increase in dark cell number in choroid plexus epithelium. Am. J. Physiol. 1999, 276, C82–C90. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Tian, G.; Leung, H.; Soo, Y.O.Y.; Chen, X.; Ip, V.H.L.; Mok, V.C.T.; Chu, W.C.W.; Wong, K.S.; Leung, T.W.H. Autonomic Dysfunction Predicts Clinical Outcomes After Acute Ischemic Stroke: A Prospective Observational Study. Stroke 2018, 49, 215–218. [Google Scholar] [CrossRef]
- Dütsch, M.; Burger, M.; Dörfler, C.; Schwab, S.; Hilz, M.J. Cardiovascular autonomic function in poststroke patients. Neurology 2007, 69, 2249–2255. [Google Scholar] [CrossRef]
- Sykora, M.; Diedler, J.; Poli, S.; Rupp, A.; Turcani, P.; Steiner, T. Blood Pressure Course in Acute Stroke Relates to Baroreflex Dysfunction. Cerebrovasc. Dis. 2010, 30, 172–179. [Google Scholar] [CrossRef]
- Sykora, M.; Diedler, J.; Turcani, P.; Hacke, W.; Steiner, T. Baroreflex: A New Therapeutic Target in Human Stroke? Stroke 2009, 40, e678–e682. [Google Scholar] [CrossRef]
- Szczepanska-Sadowska, E.; Wsol, A.; Cudnoch-Jedrzejewska, A.; Czarzasta, K.; Żera, T. Multiple Aspects of Inappropriate Action of Renin-Angiotensin, Vasopressin, and Oxytocin Systems in Neuropsychiatric and Neurodegenerative Diseases. J. Clin. Med. 2022, 11, 908. [Google Scholar] [CrossRef]
- Savić, B.; Murphy, D.; Japundžić-Žigon, N. The Paraventricular Nucleus of the Hypothalamus in Control of Blood Pressure and Blood Pressure Variability. Front. Physiol. 2022, 13, 858941. [Google Scholar] [CrossRef]
- Saravia, F.E.; Gonzalez, S.L.; Roig, P.; Alves, V.; Homo-Delarche, F.; Nicola, A.F. De Diabetes Increases the Expression of Hypothalamic Neuropeptides in a Spontaneous Model of Type I Diabetes, the Nonobese Diabetic (NOD) Mouse. Cell. Mol. Neurobiol. 2001, 21, 15–27. [Google Scholar] [CrossRef]
- Anai, H.; Ueta, Y.; Serino, R.; Nomura, M.; Kabashima, N.; Shibuya, I.; Takasugi, M.; Nakashima, Y.; Yamashita, H. Upregulation of the expression of vasopressin gene in the paraventricular and supraoptic nuclei of the lithium-induced diabetes insipidus rat. Brain Res. 1997, 772, 161–166. [Google Scholar] [CrossRef]
- Keller, W.J.; Mullaj, E. Antidiuretic hormone release associated with increased intracranial pressure independent of plasma osmolality. Brain Behav. 2018, 8, e01005. [Google Scholar] [CrossRef]
- Hacke, W.; Schwab, S.; Horn, M.; Spranger, M.; De Georgia, M.; von Kummer, R. “Malignant” middle cerebral artery territory infarction: Clinical course and prognostic signs. Arch. Neurol. 1996, 53, 309–315. [Google Scholar] [CrossRef]
- Murtha, L.A.; McLeod, D.D.; Pepperall, D.; McCann, S.K.; Beard, D.J.; Tomkins, A.J.; Holmes, W.M.; McCabe, C.; Macrae, I.M.; Spratt, N.J. Intracranial pressure elevation after ischemic stroke in rats: Cerebral edema is not the only cause, and short-duration mild hypothermia is a highly effective preventive therapy. J. Cereb. blood flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 592–600. [Google Scholar] [CrossRef] [Green Version]
- Zaghmi, A.; Dopico-López, A.; Pérez-Mato, M.; Iglesias-Rey, R.; Hervella, P.; Greschner, A.A.; Bugallo-Casal, A.; da Silva, A.; Gutiérrez-Fernández, M.; Castillo, J.; et al. Sustained blood glutamate scavenging enhances protection in ischemic stroke. Commun. Biol. 2020, 3, 729. [Google Scholar] [CrossRef]
- Dávalos, A.; Shuaib, A.; Wahlgren, N.G. Neurotransmitters and pathophysiology of stroke: Evidence for the release of glutamate and other transmitters/mediators in animals and humans. J. stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2000, 9, 2–8. [Google Scholar] [CrossRef]
- Meng, X.; Li, N.; Guo, D.-Z.; Pan, S.-Y.; Li, H.; Yang, C. High plasma glutamate levels are associated with poor functional outcome in acute ischemic stroke. Cell. Mol. Neurobiol. 2015, 35, 159–165. [Google Scholar] [CrossRef]
- Busnardo, C.; Crestani, C.C.; Resstel, L.B.M.; Tavares, R.F.; Antunes-Rodrigues, J.; Corrêa, F.M.A. Ionotropic glutamate receptors in hypothalamic paraventricular and supraoptic nuclei mediate vasopressin and oxytocin release in unanesthetized rats. Endocrinology 2012, 153, 2323–2331. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Wallace, B.K.; Yuen, N.; Jenkins, D.P.; Wulff, H.; O’Donnell, M.E. Blood-brain barrier KCa3.1 channels: Evidence for a role in brain Na uptake and edema in ischemic stroke. Stroke 2015, 46, 237–244. [Google Scholar] [CrossRef]
- Sato-Numata, K.; Numata, T.; Ueta, Y.; Okada, Y. Vasopressin Neurons Respond to Hyperosmotic Stimulation with Regulatory Volume Increase and Secretory Volume Decrease by Activating Ion Transporters and Ca(2+) Channels. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2021, 55, 119–134. [Google Scholar] [CrossRef]
- Wang, S.C.; Parpura, V.; Wang, Y.-F. Astroglial Regulation of Magnocellular Neuroendocrine Cell Activities in the Supraoptic Nucleus. Neurochem. Res. 2021, 46, 2586–2600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Parpura, V. Central Role of Maladapted Astrocytic Plasticity in Ischemic Brain Edema Formation. Front. Cell. Neurosci. 2016, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chen, M.; Wang, J.; Guo, G.; Zheng, Y.; Jiang, X.; Zhang, M. Relationship between glutamate in the limbic system and hypothalamus-pituitary-adrenal axis after middle cerebral artery occlusion in rats. Chin. Med. J. 2003, 116, 1492–1496. [Google Scholar] [PubMed]
- Cui, D.; Jia, S.; Li, T.; Li, D.; Wang, X.; Liu, X.; Wang, Y.F.; Cui, D.; Jia, S.; Liu, X.; et al. Involvement of Supraoptic Astrocytes in Basilar Artery Occlusion-Evoked Differential Activation of Vasopressin Neurons and Vasopressin Secretion in Rats. Neurochem. Res. 2021, 46, 2651–2661. [Google Scholar] [CrossRef]
- Cui, D.; Jia, S.; Li, T.; Li, D.; Wang, X.; Liu, X.; Wang, Y.F. Alleviation of brain injury by applying TGN-020 in the supraoptic nucleus via inhibiting vasopressin neurons in rats of focal ischemic stroke. Life Sci. 2021, 264, 118683. [Google Scholar] [CrossRef]
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851. [Google Scholar] [CrossRef] [Green Version]
- Szmydynger-Chodobska, J.; Fox, L.M.; Lynch, K.M.; Zink, B.J.; Chodobski, A.; Cui, D.; Jia, S.; Li, T.; Li, D.; Wang, X.; et al. Vasopressin amplifies the production of proinflammatory mediators in traumatic brain injury. J. Neurotrauma 2010, 27, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Pascale, C.L.; Szmydynger-Chodobska, J.; Sarri, J.E.; Chodobski, A. Traumatic brain injury results in a concomitant increase in neocortical expression of vasopressin and its V1a receptor. J. Physiol. Pharmacol. an Off. J. Polish Physiol. Soc. 2006, 57 (Suppl 1), 161–167. [Google Scholar]
- Liamis, G.; Barkas, F.; Megapanou, E.; Christopoulou, E.; Makri, A.; Makaritsis, K.; Ntaios, G.; Elisaf, M.; Milionis, H. Hyponatremia in Acute Stroke Patients: Pathophysiology, Clinical Significance, and Management Options. Eur. Neurol. 2020, 82, 32–40. [Google Scholar] [CrossRef]
- Robertson, G.L. Regulation of Arginine Vasopressin in the Syndrome of Inappropriate Antidiuresis. Am. J. Med. 2006, 119, 36–42. [Google Scholar] [CrossRef]
- Hannon, M.J.; Thompson, C.J. The syndrome of inappropriate antidiuretic hormone: Prevalence, causes and consequences. Eur. J. Endocrinol. 2010, 162, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, C.; Peri, A. Effects of hyponatremia on the brain. J. Clin. Med. 2014, 3, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Largeau, B.; Le Tilly, O.; Sautenet, B.; Salmon Gandonnière, C.; Barin-Le Guellec, C.; Ehrmann, S. Arginine Vasopressin and Posterior Reversible Encephalopathy Syndrome Pathophysiology: The Missing Link? Mol. Neurobiol. 2019, 56, 6792–6806. [Google Scholar] [CrossRef]
- Huang, W.-Y.; Weng, W.-C.; Peng, T.-I.; Chien, Y.-Y.; Wu, C.-L.; Lee, M.; Hung, C.-C.; Chen, K.-H. Association of Hyponatremia in Acute Stroke Stage with Three-Year Mortality in Patients with First-Ever Ischemic Stroke. Cerebrovasc. Dis. 2012, 34, 55–62. [Google Scholar] [CrossRef]
- Rodrigues, B.; Staff, I.; Fortunato, G.; McCullough, L.D. Hyponatremia in the prognosis of acute ischemic stroke. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2014, 23, 850–854. [Google Scholar] [CrossRef]
- Soiza, R.L.; Cumming, K.; Clark, A.B.; Bettencourt-Silva, J.H.; Metcalf, A.K.; Bowles, K.M.; Potter, J.F.; Myint, P.K. Hyponatremia predicts mortality after stroke. Int. J. Stroke Off. J. Int. Stroke Soc. 2015, 10 (Suppl A), 50–55. [Google Scholar] [CrossRef] [Green Version]
- Carcel, C.; Sato, S.; Zheng, D.; Heeley, E.; Arima, H.; Yang, J.; Wu, G.; Chen, G.; Zhang, S.; Delcourt, C.; et al. Prognostic Significance of Hyponatremia in Acute Intracerebral Hemorrhage: Pooled Analysis of the Intensive Blood Pressure Reduction in Acute Cerebral Hemorrhage Trial Studies. Crit. Care Med. 2016, 44, 1388–1394. [Google Scholar] [CrossRef]
- Kalita, J.; Singh, R.K.; Misra, U.K. Cerebral Salt Wasting Is the Most Common Cause of Hyponatremia in Stroke. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2017, 26, 1026–1032. [Google Scholar] [CrossRef]
- Hannon, M.J.; Behan, L.A.; O’Brien, M.M.C.; Tormey, W.; Ball, S.G.; Javadpour, M.; Sherlock, M.; Thompson, C.J. Hyponatremia following mild/moderate subarachnoid hemorrhage is due to SIAD and glucocorticoid deficiency and not cerebral salt wasting. J. Clin. Endocrinol. Metab. 2014, 99, 291–298. [Google Scholar] [CrossRef]
- Oikawa, R.; Hosoda, C.; Nasa, Y.; Daicho, T.; Tanoue, A.; Tsujimoto, G.; Takagi, N.; Tanonaka, K.; Takeo, S. Decreased susceptibility to salt-induced hypertension in subtotally nephrectomized mice lacking the vasopressin V1a receptor. Cardiovasc. Res. 2010, 87, 187–194. [Google Scholar] [CrossRef] [PubMed]
- King, K.A.; Mackie, G.; Pang, C.C.; Wall, R.A. Central vasopressin in the modulation of catecholamine release in conscious rats. Can. J. Physiol. Pharmacol. 1985, 63, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Murakami, Y.; Nakamura, K.; Yokotani, K. Vasopressin V(1) receptor-mediated activation of central sympatho-adrenomedullary outflow in rats. Eur. J. Pharmacol. 2002, 457, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Winklewski, P.J.; Radkowski, M.; Demkow, U. Cross-talk between the inflammatory response, sympathetic activation and pulmonary infection in the ischemic stroke. J. Neuroinflamm. 2014, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Shin, J. Il Inflammation and hyponatremia: An underrecognized condition? Korean J. Pediatr. 2013, 56, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Chikanza, I.C.; Grossman, A.S. Hypothalamic-pituitary-mediated immunomodulation: Arginine vasopressin is a neuroendocrine immune mediator. Br. J. Rheumatol. 1998, 37, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.H.; Spencer, R.L.; McEwen, B.S.; Stein, M. Depression, adrenal steroids, and the immune system. Ann. Med. 1993, 25, 481–487. [Google Scholar] [CrossRef]
- De Winter, R.F.P.; van Hemert, A.M.; DeRijk, R.H.; Zwinderman, K.H.; Frankhuijzen-Sierevogel, A.C.; Wiegant, V.M.; Goekoop, J.G. Anxious–Retarded Depression: Relation with Plasma Vasopressin and Cortisol. Neuropsychopharmacology 2003, 28, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Roper, J.; O’Carroll, A.-M.; Young, W., 3rd; Lolait, S. The vasopressin Avpr1b receptor: Molecular and pharmacological studies. Stress 2011, 14, 98–115. [Google Scholar] [CrossRef]
- Barugh, A.J.; Gray, P.; Shenkin, S.D.; MacLullich, A.M.J.; Mead, G.E. Cortisol levels and the severity and outcomes of acute stroke: A systematic review. J. Neurol. 2014, 261, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Geerlings, M.I.; Sigurdsson, S.; Eiriksdottir, G.; Garcia, M.E.; Harris, T.B.; Gudnason, V.; Launer, L.J. Salivary cortisol, brain volumes, and cognition in community-dwelling elderly without dementia. Neurology 2015, 85, 976–983. [Google Scholar] [CrossRef] [Green Version]
- Lupien, S.J.; de Leon, M.; de Santi, S.; Convit, A.; Tarshish, C.; Nair, N.P.; Thakur, M.; McEwen, B.S.; Hauger, R.L.; Meaney, M.J. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat. Neurosci. 1998, 1, 69–73. [Google Scholar] [CrossRef]
- Suri, D.; Vaidya, V.A. Glucocorticoid regulation of brain-derived neurotrophic factor: Relevance to hippocampal structural and functional plasticity. Neuroscience 2013, 239, 196–213. [Google Scholar] [CrossRef]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef]
- Ferris, C.F.; Melloni Jr, R.H.; Koppel, G.; Perry, K.W.; Fuller, R.W.; Delville, Y. Vasopressin/Serotonin Interactions in the Anterior Hypothalamus Control Aggressive Behavior in Golden Hamsters. J. Neurosci. 1997, 17, 4331–4340. [Google Scholar] [CrossRef] [Green Version]
- Coccaro, E.F.; Kavoussi, R.J.; Hauger, R.L.; Cooper, T.B.; Ferris, C.F. Cerebrospinal Fluid Vasopressin Levels: Correlates With Aggression and Serotonin Function in Personality-Disordered Subjects. Arch. Gen. Psychiatry 1998, 55, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.O.; Caeiro, L.; Ferro, J.M.; Albuquerque, R.; Figueira, M.L. Denial in the first days of acute stroke. J. Neurol. 2006, 253, 1016–1023. [Google Scholar] [CrossRef]
- Filep, J.; Rosenkranz, B. Mechanism of vasopressin-induced platelet aggregation. Thromb Res. 1987, 45, 7–15. [Google Scholar] [CrossRef]
- Wun, T. Vasopressin and platelets: A concise review. Platelets 1997, 8, 15–22. [Google Scholar] [CrossRef]
- Zheng, H.; Chen, C.; Zhang, J.; Hu, Z. Mechanism and therapy of brain edema after intracerebral hemorrhage. Cerebrovasc. Dis. 2016, 42, 155–169. [Google Scholar] [CrossRef]
- Szczygielski, J.; Kopańska, M.; Wysocka, A.; Oertel, J. Cerebral Microcirculation, Perivascular Unit, and Glymphatic System: Role of Aquaporin-4 as the Gatekeeper for Water Homeostasis. Front. Neurol. 2021, 12, 767470. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmelova, M.; Sucha, P.; Bochin, M.; Vorisek, I.; Pivonkova, H.; Hermanova, Z.; Anderova, M.; Vargova, L. The role of aquaporin-4 and transient receptor potential vaniloid isoform 4 channels in the development of cytotoxic edema and associated extracellular diffusion parameter changes. Eur. J. Neurosci. 2019, 50, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Hirt, L.; Fukuda, A.M.; Ambadipudi, K.; Rashid, F.; Binder, D.; Verkman, A.; Ashwal, S.; Obenaus, A.; Badaut, J. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J. Cereb. blood flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 277–290. [Google Scholar] [CrossRef]
- Yao, X.; Derugin, N.; Manley, G.T.; Verkman, A.S. Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci. Lett. 2015, 584, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Tang, Y.; Dong, Q. Protection of Vascular Endothelial Growth Factor to Brain Edema Following Intracerebral Hemorrhage and Its Involved Mechanisms: Effect of Aquaporin-4. PLoS ONE 2013, 8, e66051. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-D.; Chen, C.-C.V.; Shen, C.-C.; Chin, L.-T.; Ma, H.-I.; Chuang, H.-Y.; Cho, D.-Y.; Chu, C.-H.; Chang, C. Hyperglycemia exacerbates intracerebral hemorrhage via the downregulation of aquaporin-4: Temporal assessment with magnetic resonance imaging. Stroke 2013, 44, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Qiu, G.-P.; Xu, J.; Zhuo, F.; Sun, S.-Q.; Liu, H.; Yang, M.; Huang, J.; Lu, W.-T.; Huang, S.-Q. Loss of AQP4 polarized localization with loss of β-dystroglycan immunoreactivity may induce brain edema following intracerebral hemorrhage. Neurosci. Lett. 2015, 588, 42–48. [Google Scholar] [CrossRef]
- Moeller, H.B.; Fenton, R.A.; Zeuthen, T.; MacAulay, N. Vasopressin-dependent short-term regulation of aquaporin 4 expressed in Xenopus oocytes. Neuroscience 2009, 164, 1674–1684. [Google Scholar] [CrossRef]
- Liu, X.; Nakayama, S.; Amiry-Moghaddam, M.; Ottersen, O.P.; Bhardwaj, A. Arginine-vasopressin V1 but not V2 receptor antagonism modulates infarct volume, brain water content, and aquaporin-4 expression following experimental stroke. Neurocrit. Care 2010, 12, 124–131. [Google Scholar] [CrossRef]
- Manaenko, A.; Fathali, N.; Khatibi, N.H.; Lekic, T.; Hasegawa, Y.; Martin, R.; Tang, J.; Zhang, J.H. Arginine-vasopressin V1a receptor inhibition improves neurologic outcomes following an intracerebral hemorrhagic brain injury. Neurochem. Int. 2011, 58, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Yuen, N.; Lam, T.I.; Wallace, B.K.; Klug, N.R.; Anderson, S.E.; O’Donnell, M.E. Ischemic factor-induced increases in cerebral microvascular endothelial cell Na/H exchange activity and abundance: Evidence for involvement of ERK1/2 MAP kinase. Am. J. Physiol. Physiol. 2014, 306, C931–C942. [Google Scholar] [CrossRef] [Green Version]
- Wallace, B.K.; Foroutan, S.; O’Donnell, M.E. Ischemia-induced stimulation of Na-K-Cl cotransport in cerebral microvascular endothelial cells involves AMP kinase. Am. J. Physiol. Physiol. 2011, 301, C316–C326. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Xu, J.; Du, T.; Yan, E.; Hertz, L.; Walz, W.; Peng, L. Inhibition of brain swelling after ischemia-reperfusion by β-adrenergic antagonists: Correlation with increased K+ and decreased Ca2+ concentrations in extracellular fluid. Biomed Res. Int. 2014, 2014, 873590. [Google Scholar] [CrossRef]
- Yan, Y.; Dempsey, R.J.; Flemmer, A.; Forbush, B.; Sun, D. Inhibition of Na+–K+–Cl− cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003, 961, 22–31. [Google Scholar] [CrossRef]
- Faraco, G.; Wijasa, T.S.; Park, L.; Moore, J.; Anrather, J.; Iadecola, C. Water deprivation induces neurovascular and cognitive dysfunction through vasopressin-induced oxidative stress. J. Cereb. Blood Flow Metab. 2014, 34, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, L.A.; Wetzel, M.; Rosenberg, G.A. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005, 50, 329–339. [Google Scholar] [CrossRef]
- Wang, X.; Jung, J.; Asahi, M.; Chwang, W.; Russo, L.; Moskowitz, M.A.; Dixon, C.E.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 7037–7042. [Google Scholar] [CrossRef]
- Harkness, K.A.; Adamson, P.; Sussman, J.D.; Davies-Jones, G.A.B.; Greenwood, J.; Woodroofe, M.N. Dexamethasone regulation of matrix metalloproteinase expression in CNS vascular endothelium. Brain 2000, 123, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Tahara, A.; Tsukada, J.; Tomura, Y.; Yatsu, T.; Shibasaki, M. Vasopressin regulates rat mesangial cell growth by inducing autocrine secretion of vascular endothelial growth factor. J. Physiol. Sci. 2011, 61, 115–122. [Google Scholar] [CrossRef]
- Alonso, G.; Gallibert, E.; Lafont, C.; Guillon, G. Intrahypothalamic angiogenesis induced by osmotic stimuli correlates with local hypoxia: A potential role of confined vasoconstriction induced by dendritic secretion of vasopressin. Endocrinology 2008, 149, 4279–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bele, S. Vasopressin Increases Cerebral Perfusion Pressure but not Cerebral Blood Flow in Neurosurgical Patients with Catecholamine-Refractory Hypotension: A Preliminary Evaluation Using the Non-Invasive Quantix ND in Comparison to the Literature. J. Anesth. &Critical Care Open Access 2014, 1, 17–22. [Google Scholar] [CrossRef]
- Engelborghs, K.; Haseldonckx, M.; Van Reempts, J.; Van Rossem, K.; Wouters, L.; Borgers, M.; Verlooy, J. Impaired autoregulation of cerebral blood flow in an experimental model of traumatic brain injury. J. Neurotrauma 2000, 17, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, O.Y.; Johnson, D.; Hayes, R.L. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J. Neurotrauma 2014, 31, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Aries, M.J.H.; Elting, J.W.; De Keyser, J.; Kremer, B.P.H.; Vroomen, P.C.A.J. Cerebral autoregulation in stroke: A review of transcranial doppler studies. Stroke 2010, 41, 2697–2704. [Google Scholar] [CrossRef]
- Yamagata, K.; Sone, N.; Suguyama, S.; Nabika, T. Different effects of arginine vasopressin on high-mobility group box 1 expression in astrocytes isolated from stroke-prone spontaneously hypertensive rats and congenic SHRpch1_18 rats. Int. J. Exp. Pathol. 2016, 97, 97–106. [Google Scholar] [CrossRef]
- Lei, C.; Lin, S.; Zhang, C.; Tao, W.; Dong, W.; Hao, Z.; Liu, M.; Wu, B. High-mobility group box1 protein promotes neuroinflammation after intracerebral hemorrhage in rats. Neuroscience 2013, 228, 190–199. [Google Scholar] [CrossRef]
- Sun, S.Z.; Cao, H.; Yao, N.; Zhao, L.L.; Zhu, X.F.; Ni, E.A.; Zhu, Q.; Zhu, W. zhong β-Arrestin 2 mediates arginine vasopressin-induced IL-6 induction via the ERK1/2-NF-κB signal pathway in murine hearts. Acta Pharmacol. Sin. 2020, 41, 198–207. [Google Scholar] [CrossRef]
- Kleindienst, A.; Dunbar, J.G.; Glisson, R.; Marmarou, A. The role of vasopressin V1A receptors in cytotoxic brain edema formation following brain injury. Acta Neurochir. 2013, 155, 151–164. [Google Scholar] [CrossRef]
- Ribeiro, M.d.C.; Hirt, L.; Bogousslavsky, J.; Regli, L.; Badaut, J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J. Neurosci. Res. 2006, 83, 1231–1240. [Google Scholar] [CrossRef]
- Rutkowsky, J.M.; Wallace, B.K.; Wise, P.M.; O’Donnell, M.E. Effects of estradiol on ischemic factor-induced astrocyte swelling and AQP4 protein abundance. Am. J. Physiol. Cell Physiol. 2011, 301, C204–C212. [Google Scholar] [CrossRef]
- Frydenlund, D.S.; Bhardwaj, A.; Otsuka, T.; Mylonakou, M.N.; Yasumura, T.; Davidson, K.G.V.; Zeynalov, E.; Skare, O.; Laake, P.; Haug, F.-M.; et al. Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 13532–13536. [Google Scholar] [CrossRef] [Green Version]
- Fernández, N.; Martínez, M.A.; Luis García-Villalón, A.L.; Monge, L.; Diéguez, G. Cerebral vasoconstriction produced by vasopressin in conscious goats: Role of vasopressin V1 and V2 receptors and nitric oxide. Br. J. Pharmacol. 2001, 132, 1837–1844. [Google Scholar] [CrossRef] [Green Version]
- Van Haren, R.M.; Thorson, C.M.; Ogilvie, M.P.; Valle, E.J.; Guarch, G.A.; Jouria, J.A.; Busko, A.M.; Harris, L.T.; Bullock, M.R.; Jagid, J.R.; et al. Vasopressin for cerebral perfusion pressure management in patients with severe traumatic brain injury: Preliminary results of a randomized controlled trial. J. Trauma Acute Care Surg. 2013, 75, 1024–1030. [Google Scholar] [CrossRef]
- Onoue, H.; Kaito, N.; Tomii, M.; Tokudome, S.; Nakajima, M.; Abe, T. Human basilar and middle cerebral arteries exhibit endothelium-dependent responses to peptides. Am. J. Physiol.-Hear. Circ. Physiol. 1994, 267, H880–H886. [Google Scholar] [CrossRef]
- Takayasu, M.; Kajita, Y.; Suzuki, Y.; Shibuya, M.; Sugita, K.; Ishikawa, T.; Hidaka, H. Triphasic response of rat intracerebral arterioles to increasing concentrations of vasopressin in vitro. J. Cereb. Blood Flow Metab. 1993, 13, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Brillault, J.; Lam, T.I.; Rutkowsky, J.M.; Foroutan, S.; O’Donnell, M.E. Hypoxia effects on cell volume and ion uptake of cerebral microvascular endothelial cells. Am. J. Physiol.-Cell Physiol. 2008, 294, 88–96. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.E.; Duong, V.; Suvatne, J.; Foroutan, S.; Johnson, D.M. Arginine vasopressin stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransporter activity is V1 receptor and [Ca] dependent. Am. J. Physiol.-Cell Physiol. 2005, 289, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.K.; Tse, F.W.; Tse, A. Arginine vasopressin potentiates the stimulatory action of CRH on pituitary corticotropes via a protein kinase C-dependent reduction of the background TREK-1 current. Endocrinology 2015, 156, 3661–3672. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.; Olsson, T.; Carlberg, B.; Karlsson, K.; Fagerlund, M. Hypercortisolism after stroke--partly cytokine-mediated? J. Neurol. Sci. 1997, 147, 43–47. [Google Scholar] [CrossRef]
- Mavani, G.P.; DeVita, M.V.; Michelis, M.F. A Review of the Nonpressor and Nonantidiuretic Actions of the Hormone Vasopressin. Front. Med. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katan, M.; Fluri, F.; Morgenthaler, N.G.; Schuetz, P.; Zweifel, C.; Bingisser, R.; Müller, K.; Meckel, S.; Gass, A.; Kappos, L.; et al. Copeptin: A novel, independent prognostic marker in patients with ischemic stroke. Ann. Neurol. 2009, 66, 799–808. [Google Scholar] [CrossRef] [PubMed]
- De Marchis, G.M.; Dankowski, T.; König, I.R.; Fladt, J.; Fluri, F.; Gensicke, H.; Foerch, C.; Findling, O.; Kurmann, R.; Fischer, U.; et al. A novel biomarker-based prognostic score in acute ischemic stroke: The CoRisk score. Neurology 2019, 92, E1517–E1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The CoRisk Score Online Calculator. Available online: https://stroke-outcome.github.io/the-corisk-score/ (accessed on 10 January 2022).
- Katan, M.; Nigro, N.; Fluri, F.; Schuetz, P.; Morgenthaler, N.G.; Jax, F.; Meckel, S.; Gass, A.; Bingisser, R.; Steck, A.; et al. Stress hormones predict cerebrovascular re-events after transient ischemic attacks. Neurology 2011, 76, 563–566. [Google Scholar] [CrossRef]
- Fluri, F.; Morgenthaler, N.G.; Mueller, B.; Christ-Crain, M.; Katan, M. Copeptin, Procalcitonin and Routine Inflammatory Markers–Predictors of Infection after Stroke. PLoS ONE 2012, 7, e48309. [Google Scholar] [CrossRef]
- Hotter, B.; Hoffmann, S.; Ulm, L.; Montaner, J.; Bustamante, A.; Meisel, C.; Meisel, A. Inflammatory and stress markers predicting pneumonia, outcome, and etiology in patients with stroke: Biomarkers for predicting pneumonia, functional outcome, and death after stroke. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e692. [Google Scholar] [CrossRef] [Green Version]
- Hotter, B.; Hoffmann, S.; Ulm, L.; Meisel, C.; Bustamante, A.; Montaner, J.; Katan, M.; Smith, C.J.; Meisel, A. External Validation of Five Scores to Predict Stroke-Associated Pneumonia and the Role of Selected Blood Biomarkers. Stroke 2021, 52, 325–330. [Google Scholar] [CrossRef]
- Deboevere, N.; Marjanovic, N.; Sierecki, M.; Marchetti, M.; Dubocage, M.; Magimel, E.; Mimoz, O.; Guenezan, J. Value of copeptin and the S-100b protein assay in ruling out the diagnosis of stroke-induced dizziness pattern in emergency departments. Scand. J. Trauma. Resusc. Emerg. Med. 2019, 27, 72. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhang, L.; Wang, Q.; Zhao, J.; Liu, J.; Wang, G. Cerebrovascular risk factors associated with ischemic stroke in a young non-diabetic and non-hypertensive population: A retrospective case-control study. BMC Neurol. 2020, 20, 424. [Google Scholar] [CrossRef]
- Biscetti, F.; Giovannini, S.; Straface, G.; Bertucci, F.; Angelini, F.; Porreca, C.; Landolfi, R.; Flex, A. RANK/RANKL/OPG pathway: Genetic association with history of ischemic stroke in Italian population. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4574–4580. [Google Scholar]
- Fuellen, G.; Walter, U.; Henze, L.; Böhmert, J.; Palmer, D.; Lee, S.; Schmitt, C.A.; Rudolf, H.; Kowald, A. Protein Biomarkers in Blood Reflect the Interrelationships Between Stroke Outcome, Inflammation, Coagulation, Adhesion, Senescence and Cancer. Cell. Mol. Neurobiol. 2022, 1–12. [Google Scholar] [CrossRef]
- Meisel, A.; Meisel, C.; Harms, H.; Hartmann, O.; Ulm, L. Predicting Post-Stroke Infections and Outcome with Blood-Based Immune and Stress Markers. Cerebrovasc. Dis. 2012, 33, 580–588. [Google Scholar] [CrossRef]
- Manaenko, A.; Fathali, N.; Khatibi, N.H.; Lekic, T.; Shum, K.J.; Martin, R.; Zhang, J.H.; Tang, J. Post-treatment with SR49059 improves outcomes following an intracerebral hemorrhagic stroke in mice. Acta Neurochir. Suppl. 2011, 111, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Zeynalov, E.; Jones, S.M.; Seo, J.W.; Snell, L.D.; Elliott, J.P.; Ahmad, M. Arginine-vasopressin receptor blocker conivaptan reduces brain edema and blood-brain barrier disruption after experimental stroke in mice. PLoS ONE 2015, 10, e0136121. [Google Scholar] [CrossRef] [Green Version]
- Vakili, A.; Kataoka, H.; Plesnila, N. Role of arginine vasopressin V1 and V2 receptors for brain damage after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2005, 25, 1012–1019. [Google Scholar] [CrossRef]
- Li-Ng, M.; Verbalis, J.G. Conivaptan: Evidence supporting its therapeutic use in hyponatremia. Core Evid. 2009, 4, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Zeynalov, E.; Jones, S.M.; Elliott, J.P. Therapeutic time window for conivaptan treatment against stroke-evoked brain edema and blood-brain barrier disruption in mice. PLoS ONE 2017, 12, e0183985. [Google Scholar] [CrossRef] [Green Version]
- Can, B.; Oz, S.; Sahinturk, V.; Musmul, A.; Alatas, İ.O. Effects of conivaptan versus mannitol on post-ischemic brain injury and edema. Eurasian J. Med. 2019, 51, 42–48. [Google Scholar] [CrossRef]
- Corry, J.J.; Asaithambi, G.; Shaik, A.M.; Lassig, J.P.; Marino, E.H.; Ho, B.M.; Castle, A.L.; Banerji, N.; Tipps, M.E. Conivaptan for the Reduction of Cerebral Edema in Intracerebral Hemorrhage: A Safety and Tolerability Study. Clin. Drug Investig. 2020, 40, 503–509. [Google Scholar] [CrossRef]
- Hedna, V.S.; Bidari, S.; Gubernick, D.; Ansari, S.; Satriotomo, I.; Khan, A.A.; Qureshi, A.I. Treatment of stroke related refractory brain edema using mixed vasopressin antagonism: A case report and review of the literature. BMC Neurol. 2014, 14, 213. [Google Scholar] [CrossRef] [Green Version]
- Decker, D.; Collier, L.; Lau, T.; Olivera, R.; Roma, G.; Leonardo, C.; Seifert, H.; Rowe, D.; Pennypacker, K.R. The Effects of Clinically Relevant Hypertonic Saline and Conivaptan Administration on Ischemic Stroke. Acta Neurochir. Suppl. 2016, 121, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chojnowski, K.; Opiełka, M.; Gozdalski, J.; Radziwon, J.; Dańczyszyn, A.; Aitken, A.V.; Biancardi, V.C.; Winklewski, P.J. The Role of Arginine-Vasopressin in Stroke and the Potential Use of Arginine-Vasopressin Type 1 Receptor Antagonists in Stroke Therapy: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 2119. https://doi.org/10.3390/ijms24032119
Chojnowski K, Opiełka M, Gozdalski J, Radziwon J, Dańczyszyn A, Aitken AV, Biancardi VC, Winklewski PJ. The Role of Arginine-Vasopressin in Stroke and the Potential Use of Arginine-Vasopressin Type 1 Receptor Antagonists in Stroke Therapy: A Narrative Review. International Journal of Molecular Sciences. 2023; 24(3):2119. https://doi.org/10.3390/ijms24032119
Chicago/Turabian StyleChojnowski, Karol, Mikołaj Opiełka, Jacek Gozdalski, Jakub Radziwon, Aleksandra Dańczyszyn, Andrew Vieira Aitken, Vinicia Campana Biancardi, and Paweł Jan Winklewski. 2023. "The Role of Arginine-Vasopressin in Stroke and the Potential Use of Arginine-Vasopressin Type 1 Receptor Antagonists in Stroke Therapy: A Narrative Review" International Journal of Molecular Sciences 24, no. 3: 2119. https://doi.org/10.3390/ijms24032119