

Improving the Enzymatic Activity and Stability of a Lytic Polysaccharide Monooxygenase


Abstract
:1. Introduction
2. Results
2.1. Selection of BaLPMO10A Mutants through Sequence Consensus
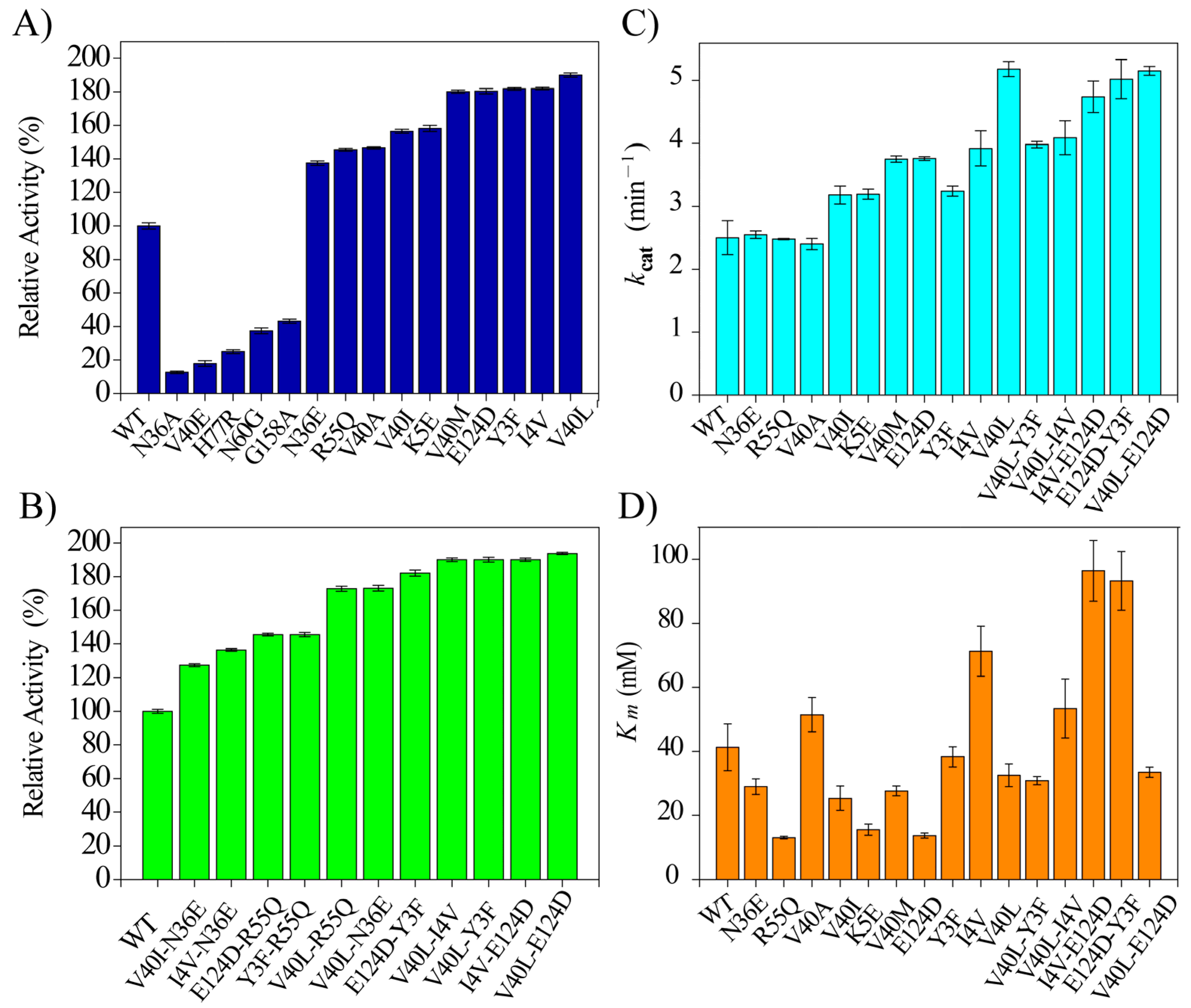
2.2. Expression and Screening of BaLPMO10A Mutants
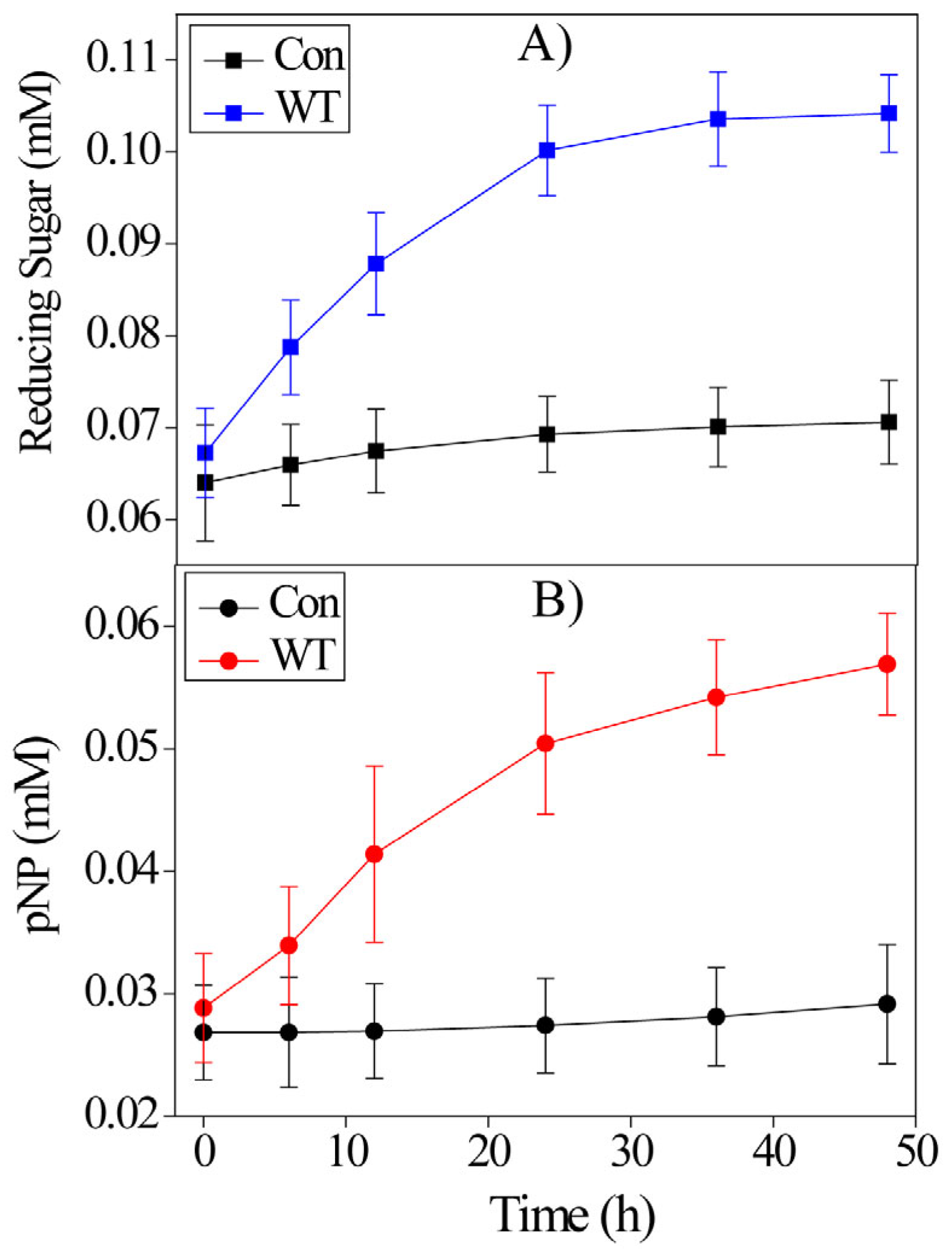
2.3. Hydrolysis Activity of BaLPMO10A
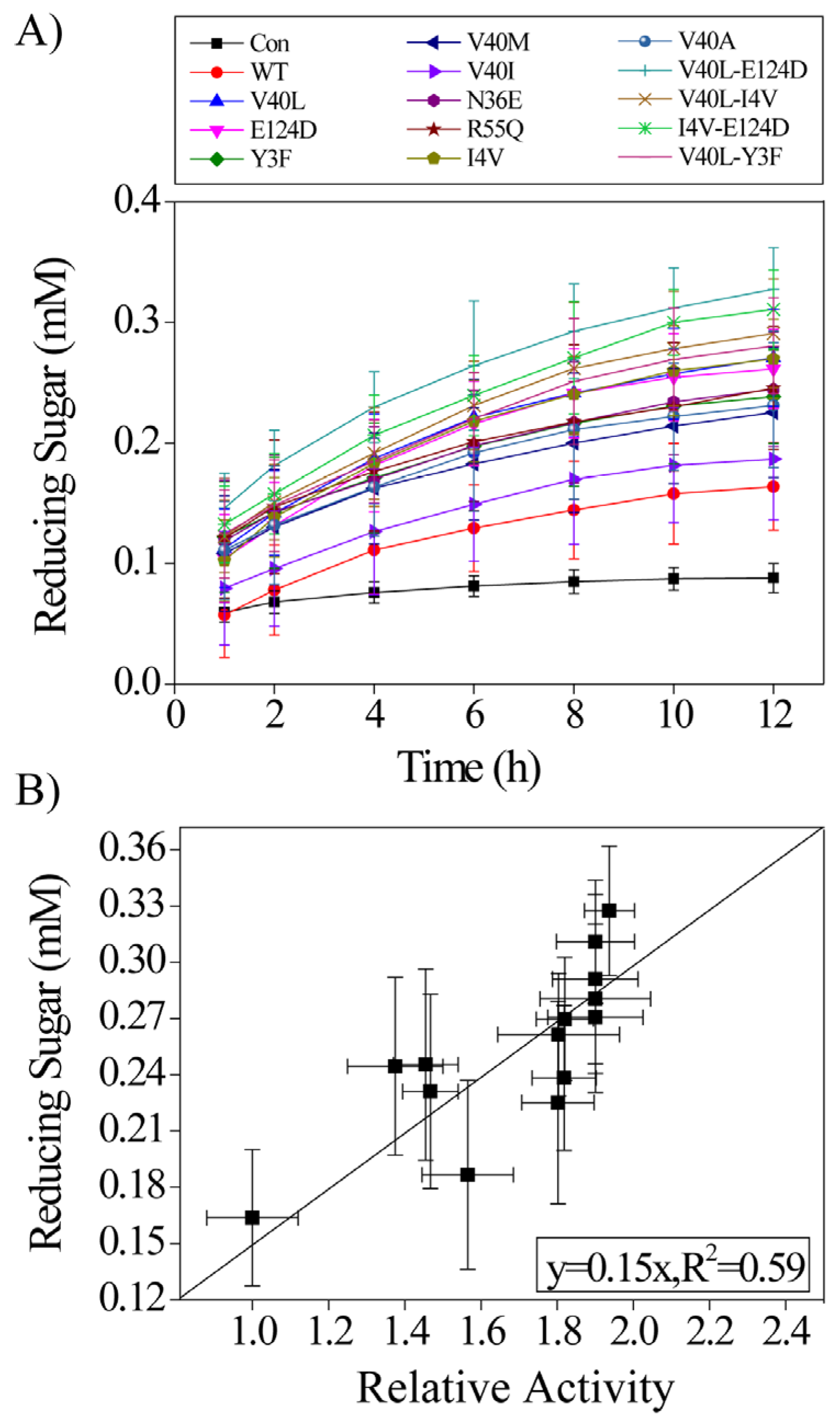
2.4. Action of the Purified BaLPMO10A Enzymes on PASC
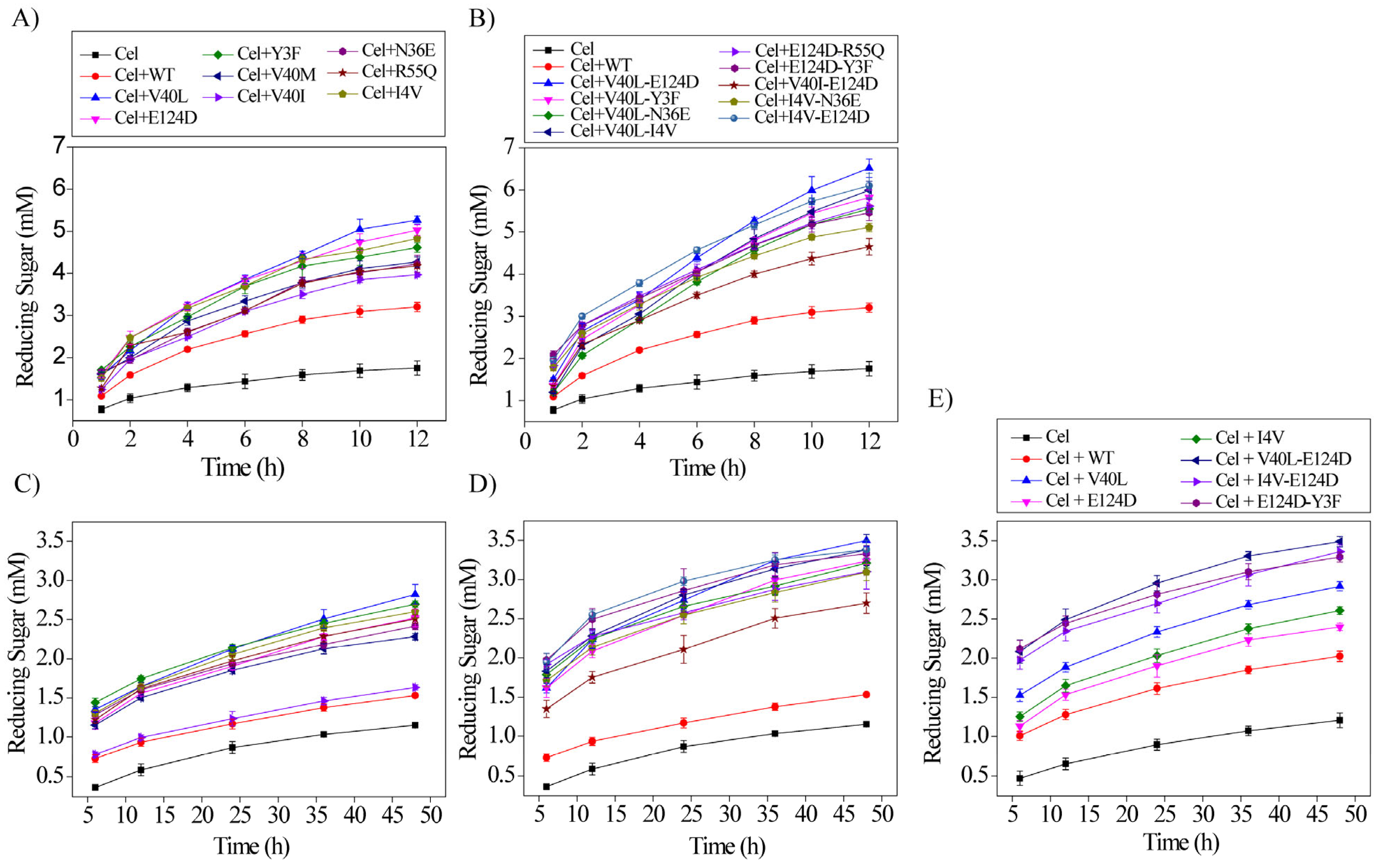
2.5. The Synergistic Depolymerization Potential of the Purified Enzymes with Cellulase against Various Substrates
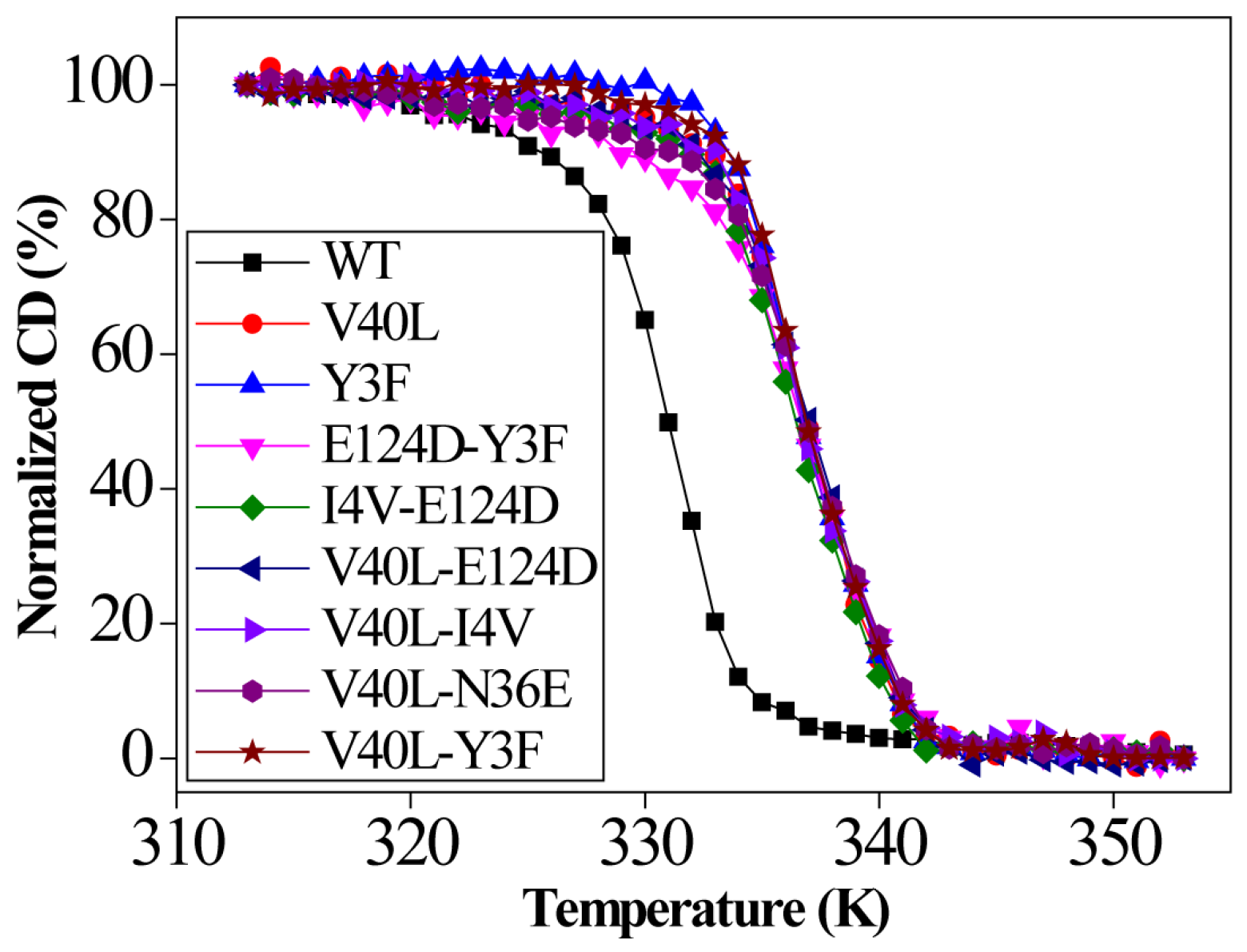
2.6. Enzyme Thermostability from CD Measurements
3. Discussion
4. Materials and Methods
4.1. Materials and Chemical Reagents
4.2. Cloning, Expression, and Purification of BaLPMO10A
4.3. Copper Saturation
4.4. Site-Directed Mutagenesis
4.5. Enzyme Activity Assay
4.6. Hydrolysis of PNPC and CMC by the Purified Enzyme
4.7. The Depolymerization of PASC by the Purified Enzymes
4.8. Synergistic Effect of the Purified Enzymes with the Commercial Cellulases on the Depolymerization of Cellulosic Substrates
4.9. Thermostability of the Purified Enzymes from CD Measurements
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | Auxiliary activity enzyme |
| CAYZy | Carbohydrate-active enzyme |
| CBM | Carbohydrate-binding module |
| CMC | Carboxymethyl cellulose |
| LPMO | Lytic polysaccharide monooxygenase |
| BaLPMO10A | LPMOs from Bacillus amyloliquefaciens |
| 2,6-DMP | 2,6-dimethoxyphenol |
| PASC | Phosphoric acid-swollen cellulose |
| FP | Filter paper |
| IPTG | Isopropyl-β-D-thiogalactopyranoside |
| H2O2 | Hydrogen peroxide |
| CD | Circular Dichroism |
| PAHBAH | Para-hydroxybenzoic acid hydrazide |
| pNPC | p-nitrophenyl-β-D-cellobioside |
| Tm | Melting temperature |
| WT | Wild type |
References
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Ezeilo, U.R.; Zakaria, I.I.; Huyop, F.; Wahab, R.A. Enzymatic breakdown of lignocellulosic biomass: The role of glycosyl hydrolases and lytic polysaccharide monooxygenases. Biotechnol. Biotechnol. Equip. 2017, 31, 647–662. [Google Scholar] [CrossRef]
- Petrovic, D.M.; Varnai, A.; Dimarogona, M.; Mathiesen, G.; Sandgren, M.; Westereng, B.; Eijsink, V.G.H. Comparison of three seemingly similar lytic polysaccharide monooxygenases from Neurospora crassa suggests different roles in plant biomass degradation. J. Biol. Chem. 2019, 294, 15068–15081. [Google Scholar] [CrossRef]
- Semenova, M.V.; Gusakov, A.V.; Telitsin, V.D.; Rozhkova, A.M.; Kondratyeva, E.G.; Sinitsyn, A.P. Purification and characterization of two forms of the homologously expressed lytic polysaccharide monooxygenase (PvLPMO9A) from Penicillium Verruculosum. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140297. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.; Chylenski, P.; Bissaro, B.; Eijsink, V.G.H.; Horn, S.J. The impact of hydrogen peroxide supply on LPMO activity and overall saccharification efficiency of a commercial cellulase cocktail. Biotechnol. Biofuels 2018, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Yang, H.; Ren, H.; Liu, R.; Sun, F.F.; Xiao, Z.; Hu, J.; Xu, Z. Synergism of Recombinant Podospora anserina PaAA9B with Cellulases Containing AA9s Can Boost the Enzymatic Hydrolysis of Cellulosic Substrates. ACS Sustain. Chem. Eng. 2020, 8, 11986–11993. [Google Scholar] [CrossRef]
- Forsberg, Z.; Mackenzie, A.K.; Sorlie, M.; Rohr, A.K.; Helland, R.; Arvai, A.S.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc. Natl. Acad. Sci. USA 2014, 111, 8446–8451. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sorlie, M.; Eijsink, V.G. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef]
- Hu, J.; Chandra, R.; Arantes, V.; Gourlay, K.; Susan van Dyk, J.; Saddler, J.N. The addition of accessory enzymes enhances the hydrolytic performance of cellulase enzymes at high solid loadings. Bioresour. Technol. 2015, 186, 149–153. [Google Scholar] [CrossRef]
- Bissaro, B.; Kommedal, E.; Rohr, A.K.; Eijsink, V.G.H. Controlled depolymerization of cellulose by light-driven lytic polysaccharide oxygenases. Nat. Commun. 2020, 11, 890. [Google Scholar] [CrossRef]
- Kracher, D.; Forsberg, Z.; Bissaro, B.; Gangl, S.; Preims, M.; Sygmund, C.; Eijsink, V.G.H.; Ludwig, R. Polysaccharide oxidation by lytic polysaccharide monooxygenase is enhanced by engineered cellobiose dehydrogenase. FEBS J. 2020, 287, 897–908. [Google Scholar] [CrossRef]
- Ciano, L.; Davies, G.J.; Tolman, W.B.; Walton, P.H. Bracing copper for the catalytic oxidation of C–H bonds. Nat. Catal. 2018, 1, 571–577. [Google Scholar] [CrossRef]
- Hemsworth, G.R.; Davies, G.J.; Walton, P.H. Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr. Opin. Struct. Biol. 2013, 23, 660–668. [Google Scholar] [CrossRef]
- Horn, S.J.; Vaaje-Kolstad, G.; Westereng, B.; Eijsink, V.G. Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 2012, 5, 45. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Chylenski, P.; Bissaro, B.; Sørlie, M.; Røhr, Å.K.; Várnai, A.; Horn, S.J.; Eijsink, V.G.H. Lytic Polysaccharide Monooxygenases in Enzymatic Processing of Lignocellulosic Biomass. ACS Catal. 2019, 9, 4970–4991. [Google Scholar] [CrossRef]
- Tandrup, T.; Frandsen, K.E.H.; Johansen, K.S.; Berrin, J.G.; Lo Leggio, L. Recent insights into lytic polysaccharide monooxygenases (LPMOs). Biochem. Soc. Trans. 2018, 46, 1431–1447. [Google Scholar] [CrossRef]
- Koseki, T.; Mese, Y.; Fushinobu, S.; Masaki, K.; Fujii, T.; Ito, K.; Shiono, Y.; Murayama, T.; Iefuji, H. Biochemical characterization of a glycoside hydrolase family 61 endoglucanase from Aspergillus kawachii. Appl. Microbiol. Biotechnol. 2008, 77, 1279–1285. [Google Scholar] [CrossRef]
- Walton, P.H.; Davies, G.J. On the catalytic mechanisms of lytic polysaccharide monooxygenases. Curr. Opin. Chem. Biol. 2016, 31, 195–207. [Google Scholar] [CrossRef]
- Quinlan, R.J.; Sweeney, M.D.; Lo Leggio, L.; Otten, H.; Poulsen, J.C.; Johansen, K.S.; Krogh, K.B.; Jorgensen, C.I.; Tovborg, M.; Anthonsen, A.; et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. USA 2011, 108, 15079–15084. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Forsberg, Z.; Loose, J.S.; Bissaro, B.; Eijsink, V.G. Structural diversity of lytic polysaccharide monooxygenases. Curr. Opin. Struct. Biol. 2017, 44, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Courtade, G.; Forsberg, Z.; Heggset, E.B.; Eijsink, V.G.H.; Aachmann, F.L. The carbohydrate-binding module and linker of a modular lytic polysaccharide monooxygenase promote localized cellulose oxidation. J. Biol. Chem. 2018, 293, 13006–13015. [Google Scholar] [CrossRef] [PubMed]
- Bissaro, B.; Røhr, Å.K.; Müller, G.; Chylenski, P.; Skaugen, M.; Forsberg, Z.; Horn, S.J.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 2017, 13, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Eijsink, V.G.H.; Petrovic, D.; Forsberg, Z.; Mekasha, S.; Rohr, A.K.; Varnai, A.; Bissaro, B.; Vaaje-Kolstad, G. On the functional characterization of lytic polysaccharide monooxygenases (LPMOs). Biotechnol. Biofuels. 2019, 12, 58. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Eggert, T. Enantioselective biocatalysis optimized by directed evolution. Curr. Opin. Biotechnol. 2004, 15, 305–313. [Google Scholar] [CrossRef]
- Martinez, A.T.; Ruiz-Duenas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef]
- Breslmayr, E.; Hanzek, M.; Hanrahan, A.; Leitner, C.; Kittl, R.; Santek, B.; Oostenbrink, C.; Ludwig, R. A fast and sensitive activity assay for lytic polysaccharide monooxygenase. Biotechnol. Biofuels 2018, 11, 79. [Google Scholar] [CrossRef]
- Breslmayr, E.; Daly, S.; Pozgajcic, A.; Chang, H.; Rezic, T.; Oostenbrink, C.; Ludwig, R. Improved spectrophotometric assay for lytic polysaccharide monooxygenase. Biotechnol. Biofuels 2019, 12, 283. [Google Scholar] [CrossRef]
- Brander, S.; Lausten, S.; Ipsen, J.O.; Falkenberg, K.B.; Bertelsen, A.B.; Norholm, M.H.H.; Ostergaard, L.H.; Johansen, K.S. Colorimetric LPMO assay with direct implication for cellulolytic activity. Biotechnol. Biofuels 2021, 14, 51. [Google Scholar] [CrossRef]
- Gregory, R.C.; Hemsworth, G.R.; Turkenburg, J.P.; Hart, S.J.; Walton, P.H.; Davies, G.J. Activity, stability and 3-D structure of the Cu(ii) form of a chitin-active lytic polysaccharide monooxygenase from Bacillus amyloliquefaciens. Dalton Trans. 2016, 45, 16904–16912. [Google Scholar] [CrossRef]
- Harris, P.V.; Welner, D.; McFarland, K.C.; Re, E.; Navarro Poulsen, J.C.; Brown, K.; Salbo, R.; Ding, H.; Vlasenko, E.; Merino, S.; et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: Structure and function of a large, enigmatic family. Biochemistry 2010, 49, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Lever, M. Colorimetric and fluorometric carbohydrate determination with p-hydroxybenzoic acid hydrazide. Biochem. Med. 1973, 7, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, Z.; Li, Y.; He, J.; Zhu, H. Improvement of the Stability and Activity of an LPMO Through Rational Disulfide Bonds Design. Front. Bioeng. Biotechnol. 2021, 9, 815990. [Google Scholar] [CrossRef] [PubMed]
- Lo Leggio, L.; Weihe, C.D.; Poulsen, J.N.; Sweeney, M.; Rasmussen, F.; Lin, J.; De Maria, L.; Wogulis, M. Structure of a lytic polysaccharide monooxygenase from Aspergillus fumigatus and an engineered thermostable variant. Carbohydr. Res. 2018, 469, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Hansson, H.; Karkehabadi, S.; Mikkelsen, N.; Douglas, N.R.; Kim, S.; Lam, A.; Kaper, T.; Kelemen, B.; Meier, K.K.; Jones, S.M.; et al. High-resolution structure of a lytic polysaccharide monooxygenase from Hypocrea jecorina reveals a predicted linker as an integral part of the catalytic domain. J. Biol. Chem. 2017, 292, 19099–19109. [Google Scholar] [CrossRef]
- Ceccherini, S.; Rahikainen, J.; Marjamaa, K.; Sawada, D.; Grönqvist, S.; Maloney, T. Activation of softwood Kraft pulp at high solids content by endoglucanase and lytic polysaccharide monooxygenase. Ind. Crop. Prod. 2021, 166, 113463. [Google Scholar] [CrossRef]
- Song, B.; Li, B.; Wang, X.; Shen, W.; Park, S.; Collings, C.; Feng, A.; Smith, S.J.; Walton, J.D.; Ding, S.Y. Real-time imaging reveals that lytic polysaccharide monooxygenase promotes cellulase activity by increasing cellulose accessibility. Biotechnol. Biofuels 2018, 11, 41. [Google Scholar] [CrossRef]
- Tokin, R.; Ipsen, J.O.; Westh, P.; Johansen, K.S. The synergy between LPMOs and cellulases in enzymatic saccharification of cellulose is both enzyme- and substrate-dependent. Biotechnol. Lett. 2020, 42, 1975–1984. [Google Scholar] [CrossRef]
- Escuder-Rodriguez, J.J.; DeCastro, M.E.; Cerdan, M.E.; Rodriguez-Belmonte, E.; Becerra, M.; Gonzalez-Siso, M.I. Cellulases from Thermophiles Found by Metagenomics. Microorganisms 2018, 6, 66. [Google Scholar] [CrossRef]
- Tanghe, M.; Danneels, B.; Last, M.; Beerens, K.; Stals, I.; Desmet, T. Disulfide bridges as essential elements for the thermostability of lytic polysaccharide monooxygenase LPMO10C from Streptomyces coelicolor. Protein Eng. Des. Sel. 2017, 30, 401–408. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, Z.; He, J.; Li, Y.; Pan, C.; Wang, C.; Deng, M.R.; Zhu, H. A myxobacterial LPMO10 has oxidizing cellulose activity for promoting biomass enzymatic saccharification of agricultural crop straws. Bioresour. Technol. 2020, 318, 124217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, S.; Yao, L. A mechanistic study of Trichoderma reesei Cel7B catalyzed glycosidic bond cleavage. J. Phys. Chem. B 2013, 117, 8714–8722. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Laurent, C.; Boerkamp, V.J.P.; van Erven, G.; Ludwig, R.; van Berkel, W.J.H.; Kabel, M.A. Regioselective C4 and C6 Double Oxidation of Cellulose by Lytic Polysaccharide Monooxygenases. Chemsuschem 2022, 15, e202102203. [Google Scholar] [CrossRef] [PubMed]
- Ghatge, S.S.; Telke, A.A.; Waghmode, T.R.; Lee, Y.; Lee, K.W.; Oh, D.B.; Shin, H.D.; Kim, S.W. Multifunctional cellulolytic auxiliary activity protein HcAA10-2 from Hahella chejuensis enhances enzymatic hydrolysis of crystalline cellulose. Appl. Microbiol. Biotechnol. 2015, 99, 3041–3055. [Google Scholar] [CrossRef]
- Claeyssens, M.; van Tilbeurgh, H.; Kamerling, J.P.; Berg, J.; Vrsanska, M.; Biely, P. Studies of the cellulolytic system of the filamentous fungus Trichoderma reesei QM 9414. Substrate specificity and transfer activity of endoglucanase I. Biochem. J. 1990, 270, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Loose, J.S.; Forsberg, Z.; Fraaije, M.W.; Eijsink, V.G.; Vaaje-Kolstad, G. A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett. 2014, 588, 3435–3440. [Google Scholar] [CrossRef]
- Wang, W.; Malcolm, B.A. Two-stage polymerase chain reaction protocol allowing introduction of multiple mutations, deletions, and insertions, using Quik Change site-directed mutagenesis. Methods. Mol. Biol. 2002, 182, 37–43. [Google Scholar]
- Shu, Z.; Wang, Y.; An, L.; Yao, L. The slowdown of the endoglucanase Trichoderma reesei Cel5A catalyzed cellulose hydrolysis is related to its initial activity. Biochemistry 2014, 53, 7650–7658. [Google Scholar] [CrossRef]
- Cannella, D.; Mollers, K.B.; Frigaard, N.U.; Jensen, P.E.; Bjerrum, M.J.; Johansen, K.S.; Felby, C. Light-driven oxidation of polysaccharides by photosynthetic pigments and a metalloenzyme. Nat. Commun. 2016, 7, 11134. [Google Scholar] [CrossRef]
- Wood, T.M. Preparation of crystalline, amorphous, and dyed cellulase substrates. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1988; Volume 160, pp. 19–25. [Google Scholar]
- Murphy, L.; Cruys-Bagger, N.; Damgaard, H.D.; Baumann, M.J.; Olsen, S.N.; Borch, K.; Lassen, S.F.; Sweeney, M.; Tatsumi, H.; Westh, P. Origin of initial burst in activity for Trichoderma reesei endo-glucanases hydrolyzing insoluble cellulose. J. Biol. Chem. 2012, 287, 1252–1260. [Google Scholar] [CrossRef]
- Singh, R.K.; Blossom, B.M.; Russo, D.A.; van Oort, B.; Croce, R.; Jensen, P.E.; Felby, C.; Bjerrum, M.J. Thermal unfolding and refolding of a lytic polysaccharide monooxygenase from Aurantiacus. RSC Adv. 2019, 9, 29734–29742. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BaLPMO10A | Tm (°C) | BaLPMO10A | Tm (°C) |
|---|---|---|---|
| WT | 57.30 ± 0.12 | V40M | 63.10 ± 0.12 |
| V40I-Y3F | 55.25 ± 0.22 | I4V-E124D | 63.80 ± 0.20 |
| V40I-E124D | 59.30 ± 0.08 | Y3F | 63.90 ± 0.22 |
| V40I-R55Q | 59.90 ± 0.20 | V40L-Y3F | 64.10 ± 0.19 |
| V40L-R55Q | 60.20 ± 0.24 | E124D | 63.14 ± 0.10 |
| E124D-R55 | 60.35 ± 0.10 | V40L-I4V | 63.90 ± 0.25 |
| N36E | 60.45 ± 0.15 | V40L | 64.20 ± 0.20 |
| Y3F-R55Q | 60.85± 0.09 | V40L-Y3F | 64.30 ± 0.28 |
| K5E | 62.60 ± 0.37 | V40L-N36E | 64.57 ± 0.22 |
| V40I | 62.80 ± 0.11 | V40L-E124D | 64.82 ± 0.20 |
| I4V | 62.90 ± 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berhe, M.H.; Song, X.; Yao, L. Improving the Enzymatic Activity and Stability of a Lytic Polysaccharide Monooxygenase. Int. J. Mol. Sci. 2023, 24, 8963. https://doi.org/10.3390/ijms24108963
Berhe MH, Song X, Yao L. Improving the Enzymatic Activity and Stability of a Lytic Polysaccharide Monooxygenase. International Journal of Molecular Sciences. 2023; 24(10):8963. https://doi.org/10.3390/ijms24108963
Chicago/Turabian StyleBerhe, Miesho Hadush, Xiangfei Song, and Lishan Yao. 2023. "Improving the Enzymatic Activity and Stability of a Lytic Polysaccharide Monooxygenase" International Journal of Molecular Sciences 24, no. 10: 8963. https://doi.org/10.3390/ijms24108963