
Microphthalmia-Associated Transcription Factor: A Differentiation Marker in Uveal Melanoma
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
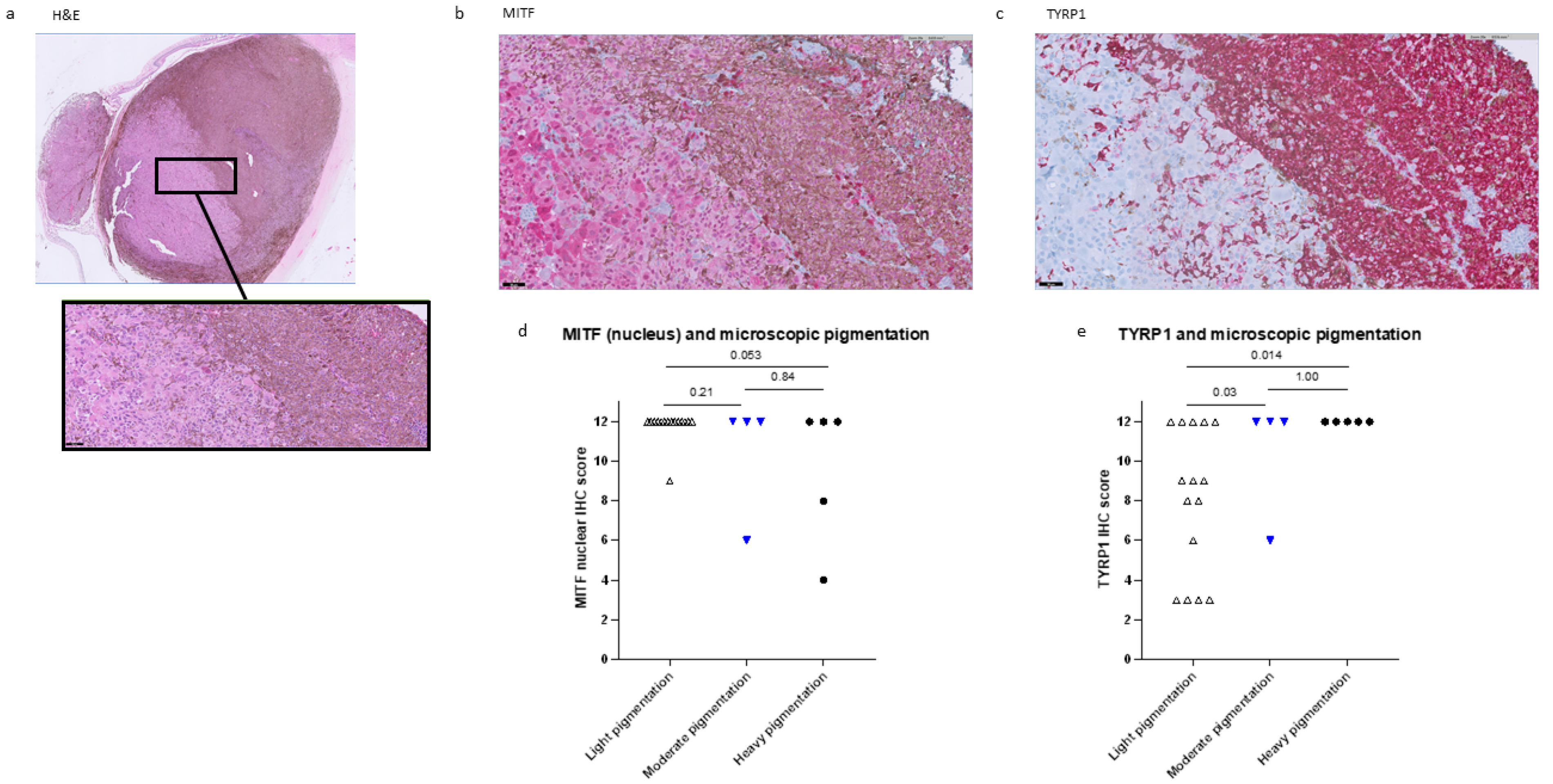
2.1. Histopathological Data
2.2. Genetic Associations
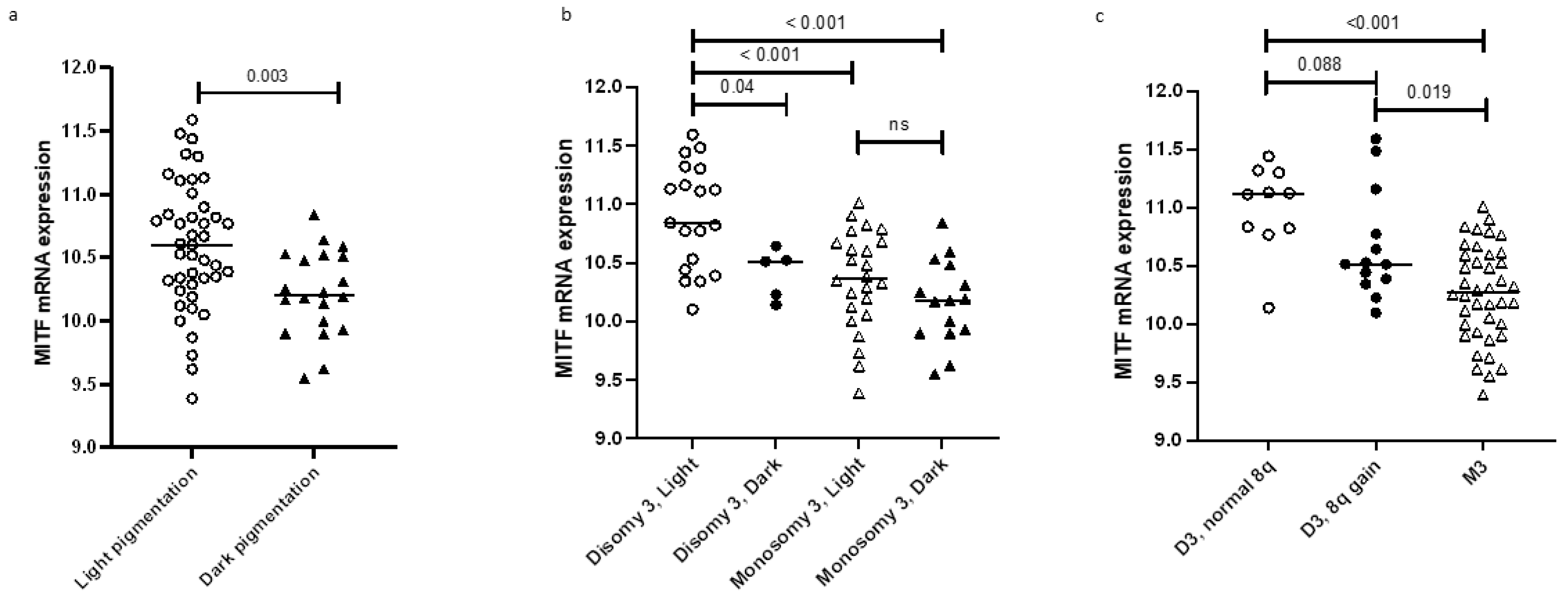
2.3. MITF Expression and Pigmentation
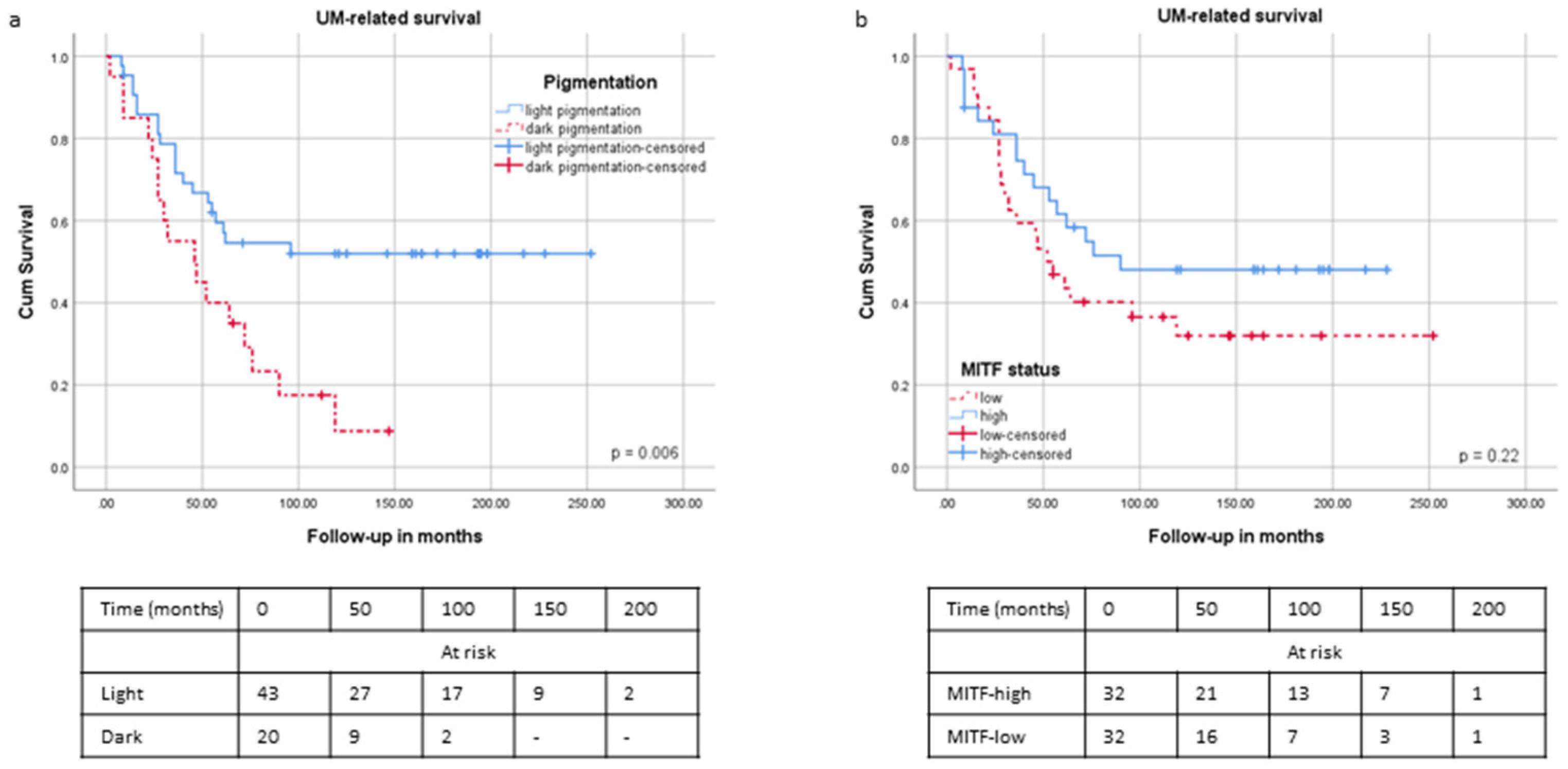
2.4. Survival and Pigmentation
2.5. Inflammation
2.6. EMT
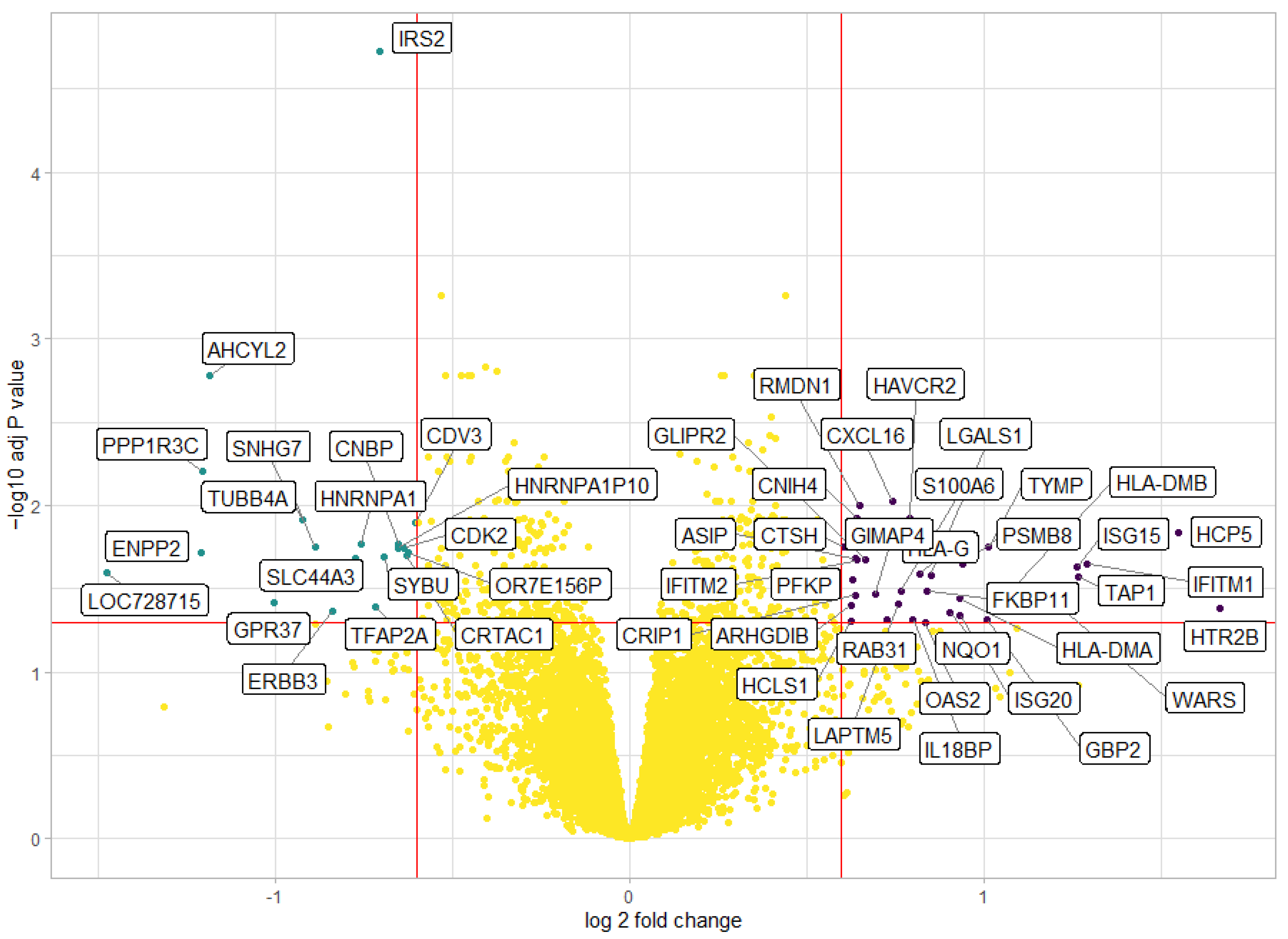
2.7. Differential Gene Expression Analysis
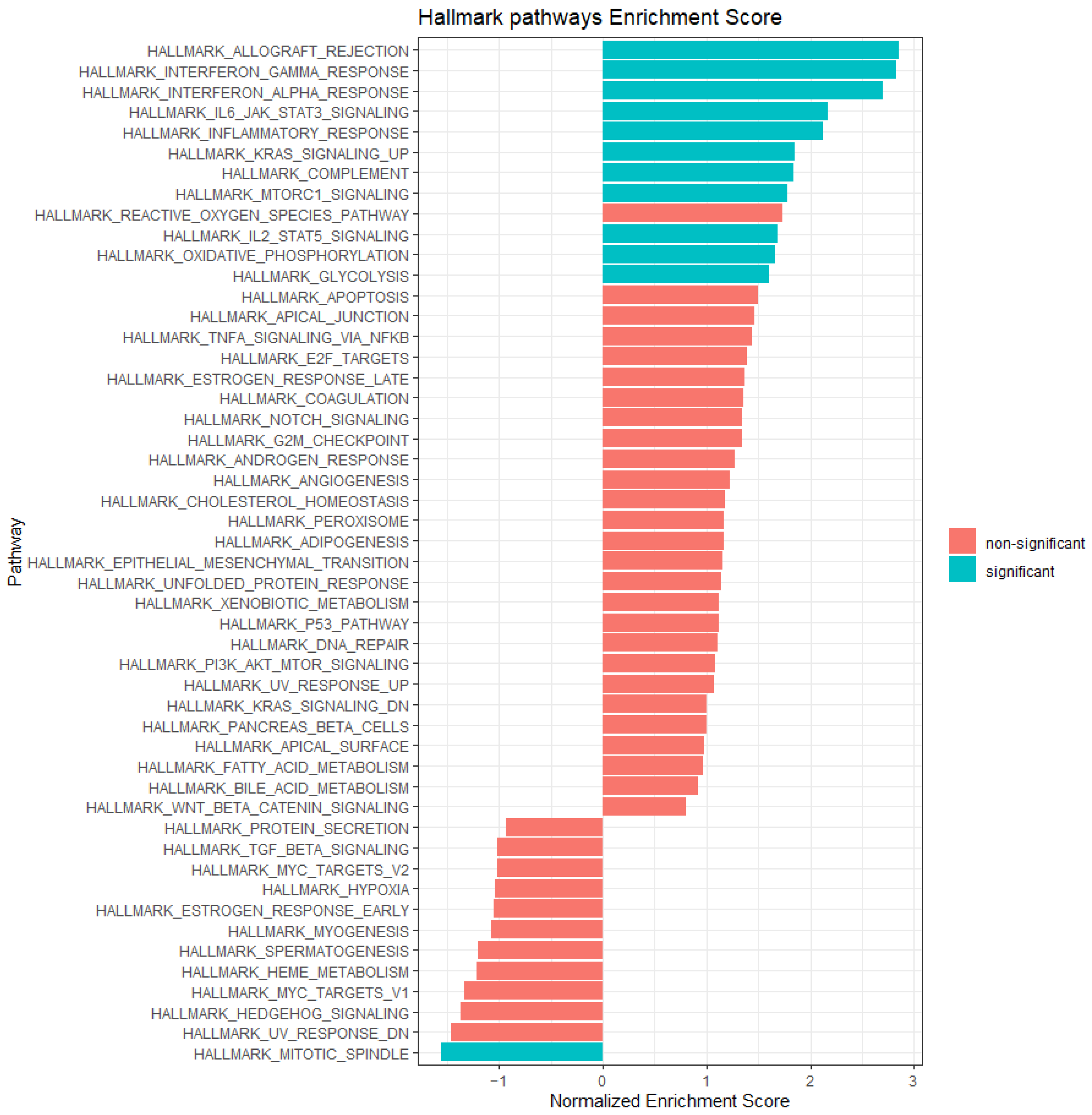
2.8. Gene Set Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Samples for Immunohistochemical (IHC) Staining
4.2. IHC Staining and Scoring
4.3. Statistical Analyses
4.4. Study Approval
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal melanoma. Nat. Rev. Dis. Prim. 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.; Furuta, M.; Thangappan, A.; Nagori, S.; Mashayekhi, A.; Lally, D.; Kelly, C.; Rudich, D.; Nagori, A.; Wakade, O.; et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch. Ophthalmol. 2009, 127, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef]
- Roelofsen, C.D.M.; Wierenga, A.P.A.; van Duinen, S.; Verdijk, R.M.; Bleeker, J.; Marinkovic, M.; Luyten, G.P.M.; Jager, M.J. Five Decades of Enucleations for Uveal Melanoma in One Center: More Tumors with High Risk Factors, No Improvement in Survival over Time. Ocul. Oncol. Pathol. 2021, 7, 133–141. [Google Scholar] [CrossRef]
- Mooy, C.M.; De Jong, P.T. Prognostic parameters in uveal melanoma: A review. Surv. Ophthalmol. 1996, 41, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, W.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Horsman, D.E.; Sroka, H.; Rootman, J.; White, V.A. Monosomy 3 and Isochromosome 8q in a Uveal Melanoma. Cancer Genet. Cytogenet. 1990, 45, 249–253. [Google Scholar] [CrossRef]
- Kilic, E.; van Gils, W.; Lodder, E.; Beverloo, H.B.; van Til, M.E.; Mooy, C.M.; Paridaens, D.; de Klein, A.; Luyten, G.P.M. Clinical and Cytogenetic Analyses in Uveal Melanoma. Investig. Opthalmol. Vis. Sci. 2006, 47, 3703–3707. [Google Scholar] [CrossRef]
- Damato, B.; Dopierala, J.; Klaasen, A.; van Dijk, M.; Sibbring, J.; Coupland, S.E. Multiplex ligation-dependent probe amplification of uveal melanoma: Correlation with metastatic death. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3048–3055. [Google Scholar] [CrossRef]
- Dogrusoz, M.; Jager, M.J. Genetic prognostication in uveal melanoma. Acta Ophthalmol. 2018, 96, 331–347. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene Expression Profiling in Uveal Melanoma Reveals Two Molecular Classes and Predicts Metastatic Death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W. A prognostic test to predict the risk of metastasis in uveal melanoma based on a 15-gene expression profile. Methods Mol. Biol. 2014, 1102, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Kaliki, S.; Cohen, M.N.; Shields, P.W.; Furuta, M.; Shields, J.A. Prognosis of uveal melanoma based on race in 8100 patients: The 2015 Doyne Lecture. Eye 2015, 29, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- McLean, M.I.W.; Foster, W.D.; Zimmerman, L.E. Prognostic Factors in Small Malignant Melanomas of Choroid and Ciliary Body. Arch. Ophthalmol. 1977, 95, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Albert, D.M.; Lavin, P.T.; Robinson, N. A Prognostic Factor Study of Disease-Free Interval and Survival Following Enucleation for Uveal Melanoma. Arch. Ophthalmol. 1983, 101, 1894–1899. [Google Scholar] [CrossRef]
- Gelmi, M.C.; Wierenga, A.P.A.; Kroes, W.G.M.; van Duinen, S.G.; Karuntu, J.S.; Marinkovic, M.; Bleeker, J.C.; Luyten, G.P.M.; Vu, T.H.K.; Verdijk, R.M.; et al. Increased histological tumour pigmentation in Uveal Melanoma is related to eye colour and loss of chromosome 3/BAP1. Ophthalmol. Sci. 2023, 3, 100297. [Google Scholar] [CrossRef]
- Gelmi, M.C.; Houtzagers, L.E.; Strub, T.; Krossa, I.; Jager, M.J. MITF in Normal Melanocytes, Cutaneous and Uveal Melanoma: A Delicate Balance. Int. J. Mol. Sci. 2022, 23, 6001. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Jung, S.-H. Downregulation of melanogenesis: Drug discovery and therapeutic options. Drug Discov. Today 2017, 22, 282–298. [Google Scholar] [CrossRef]
- Hodgkinson, C.A.; Moore, K.J.; Nakayama, A.; Steingrimsson, E.; Copeland, N.G.; Jenkins, N.; Arnheiter, H. Mutations at the Mouse Microphthalmia Locus Are Associated with Defects in a Gene Encoding a Novel Basic-Helix-Loop-Helix-Zipper Protein. Cell 1993, 74, 395–404. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Robinson, K.C.; Veguilla, R.A.; Chen, P.H.; Yokoyama, S.; Makino, E.; Lu, J.; Larue, L.; Beermann, F.; et al. Lineage-specific transcriptional regulation of DICER by MITF in melanocytes. Cell 2010, 141, 994–1005. [Google Scholar] [CrossRef]
- McGill, G.; Horstmann, M.A.; Widlund, H.R.; Nishimura, E.K.; Lin, Y.; Ramaswamy, S.; Avery, W.; Ding, H.; Jordan, S.A.; Jackson, I.J.; et al. Bcl2 Regulation by the Melanocyte Master Regulator Mitf Modulates Lineage Survival and Melanoma Cell Viability. Cell 2002, 109, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef]
- Cheli, Y.; Guiliano, S.; Botton, T.; Rocchi, S.; Hofman, V.; Hofman, P.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 2011, 30, 2307–2318. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Matatall, K.A.; Agapova, O.A.; Onken, M.D.; Worley, L.A.; Bowcock, A.M.; Harbour, J.W. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer 2013, 13, 371. [Google Scholar] [CrossRef]
- Mouriaux, F.; Vincent, S.; Kherrouche, Z.; Maurage, C.-A.; Planque, N.; Monté, D.; Labalette, P.; Saule, S. Microphthalmia transcription factor analysis in posterior uveal melanomas. Exp. Eye Res. 2003, 76, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Phelps, G.B.; Hagen, H.R.; Amsterdam, A.; Lees, J.A. MITF deficiency accelerates GNAQ-driven uveal melanoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2107006119. [Google Scholar] [CrossRef]
- Perez, D.E.; Henle, A.M.; Amsterdam, A.; Hagen, H.R.; Lees, J.A. Uveal melanoma driver mutations in GNAQ/11 yield numerous changes in melanocyte biology. Pigment Cell Melanoma Res. 2018, 31, 604–613. [Google Scholar] [CrossRef]
- Jager, M.J.; Hurks, H.M.H.; Levitskaya, J.; Kiessling, R. HLA Expression in Uveal Melanoma: There is No Rule without Some Exception. Hum. Immunol. 2002, 63, 444–451. [Google Scholar] [CrossRef]
- Bronkhorst, I.H.; Ly, L.V.; Jordanova, E.S.; Vrolijk, J.; Versluis, M.; Luyten, G.P.; Jager, M.J. Detection of M2-macrophages in uveal melanoma and relation with survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 643–650. [Google Scholar] [CrossRef]
- Ericsson, C.; Seregard, S.; Bartolazzi, A.; Levitskaya, E.; Ferrone, S.; Kiessling, R.; Larsson, O. Association of HLA Class I and Class II Antigen Expression and Mortality in Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2153–2156. [Google Scholar]
- Gezgin, G.; Dogrusoz, M.; van Essen, T.H.; Kroes, W.G.M.; Luyten, G.P.M.; van der Velden, P.A.; Walter, V.; Verdijk, R.M.; van Hall, T.; van der Burg, S.H.; et al. Genetic evolution of uveal melanoma guides the development of an inflammatory microenvironment. Cancer Immunol. Immunother. 2017, 66, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef]
- Fu, C.; Chen, J.; Lu, J.; Yi, L.; Tong, X.; Kang, L.; Pei, S.; Ouyang, Y.; Jiang, L.; Ding, Y.; et al. Roles of inflammation factors in melanogenesis (Review). Mol. Med. Rep. 2020, 21, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Paus, R.; Schadendorf, D. Melanocytes as “sensory” and regulatory cells in the epidermis. J. Theor. Biol. 1993, 164, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A. Neuroendocrine activity of the melanocyte. Exp. Dermatol. 2009, 18, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Plonka, P.M.; Passeron, T.; Brenner, M.; Tobin, D.J.; Shibahara, S.; Thomas, A.; Slominski, A.; Kadekaro, A.L.; Hershkovitz, D.; Peters, E.; et al. What are melanocytes really doing all day long...? Exp. Dermatol. 2009, 18, 799–819. [Google Scholar] [CrossRef]
- Le Poole, I.C.; Mutis, T.; van den Wijngaard, R.M.; Westerhof, W.; Ottenhoff, T.; de Vries, R.R.; Das, P.K. A novel, antigen-presenting function of melanocytes and its possible relationship to hypopigmentary disorders. J. Immunol. 1993, 151, 7284–7292. [Google Scholar] [CrossRef]
- Le Poole, I.C.; van den Wijngaard, R.M.; Westerhof, W.; Verkruisen, R.P.; Dutrieux, R.P.; Dingemans, K.P.; Das, P.K. Phagocytosis by normal human melanocytes in vitro. Exp. Cell Res. 1993, 205, 388–395. [Google Scholar] [CrossRef]
- Cioanca, V.A.; McCluskey, P.J.; Madigan, M.C. Human choroidal melanocytes express functional Toll-like receptors (TLRs). Exp. Eye Res. 2018, 173, 73–84. [Google Scholar] [CrossRef]
- Yu, N.; Zhang, S.; Zuo, F.; Kang, K.; Guan, M.; Xiang, L. Cultured human melanocytes express functional toll-like receptors 2-4, 7 and 9. J. Dermatol. Sci. 2009, 56, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, N.; Denecker, G.; Bruneel, K.; Blancke, G.; Akay, O.; Taminau, J.; De Coninck, J.; De Smedt, E.; Skrypek, N.; Van Loocke, W.; et al. The EMT Transcription Factor ZEB2 Promotes Proliferation of Primary and Metastatic Melanoma While Suppressing an Invasive, Mesenchymal-Like Phenotype. Cancer Res. 2020, 80, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Asnaghi, L.; Gezgin, G.; Tripathy, A.; Handa, J.T.; Merbs, S.L.; Van der Velden, P.A.; Jager, M.J.; Harbour, J.W.; Eberhart, C.G. EMT-associated factors promote invasive properties of uveal melanoma cells. Mol. Vis. 2015, 21, 919–929. [Google Scholar] [PubMed]
- Arts, N.; Cane, S.; Hennequart, M.; Lamy, J.; Bommer, G.; Van den Eynde, B.; De Plaen, E. microRNA-155, induced by interleukin-1ss, represses the expression of microphthalmia-associated transcription factor (MITF-M) in melanoma cells. PLoS ONE 2015, 10, e0122517. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Del-Campo, L.; Marti-Diaz, R.; Montenegro, M.F.; Gonzalez-Guerrero, R.; Hernandez-Caselles, T.; Martinez-Barba, E.; Pinero-Madrona, A.; Cabezas-Herrera, J.; Goding, C.R.; Rodriguez-Lopez, J.N. MITF induces escape from innate immunity in melanoma. J. Exp. Clin. Cancer Res. 2021, 40, 117. [Google Scholar] [CrossRef]
- Bronkhorst, I.H.; Jager, M.J. Uveal melanoma: The inflammatory microenvironment. J. Innate Immun. 2012, 4, 454–462. [Google Scholar] [CrossRef]
- Wiedemann, G.M.; Aithal, C.; Kraechan, A.; Heise, C.; Cadilha, B.L.; Zhang, J.; Duewell, P.; Ballotti, R.; Endres, S.; Bertolotto, C.; et al. Microphthalmia-Associated Transcription Factor (MITF) Regulates Immune Cell Migration into Melanoma. Transl. Oncol. 2019, 12, 350–360. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Tassabehji, M.; Newton, V.E.; Read, A.P. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat. Genet. 1994, 8, 251–255. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Weilbaecher, K.N.; McGill, G.; Cooley, E.; Mihm, M.; Fisher, D.E. Microphthalmia transcription factor. A sensitive and specific melanocyte marker for MelanomaDiagnosis. Am. J. Pathol. 1999, 155, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Granter, S.R.; Weilbaecher, K.N.; Quigley, C.; Fletcher, C.D.; Fisher, D.E. Clear cell sarcoma shows immunoreactivity for microphthalmia transcription factor: Further evidence for melanocytic differentiation. Mod. Pathol. 2001, 14, 6–9. [Google Scholar] [CrossRef]
- Granter, S.R.; Weilbaecher, K.N.; Quigley, C.; Fisher, D.E. Role for microphthalmia transcription factor in the diagnosis of metastatic malignant melanoma. Appl. Immunohistochem. Mol. Morphol. 2002, 10, 47–51. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.H.; Christopher, B.N.; Faramawi, M.F.; Said-Ahmed, K.; Cole, C.; McFaddin, A.; Ray-Chaudhury, A.; Heerema, N.; Davidorf, F.H. Frequency, molecular pathology and potential clinical significance of partial chromosome 3 aberrations in uveal melanoma. Mod. Pathol. 2011, 24, 954–962. [Google Scholar] [CrossRef]
- Rodrigues, M.; Ait Rais, K.; Salviat, F.; Algret, N.; Simaga, F.; Barnhill, R.; Gardrat, S.; Servois, V.; Mariani, P.; Piperno-Neumann, S.; et al. Association of Partial Chromosome 3 Deletion in Uveal Melanomas with Metastasis-Free Survival. JAMA Ophthalmol. 2020, 138, 182–188. [Google Scholar] [CrossRef]
- Thomas, S.; Pütter, C.; Weber, S.; Bornfeld, N.; Lohmann, D.R.; Zeschnigk, M. Prognostic significance of chromosome 3 alterations determined by microsatellite analysis in uveal melanoma: A long-term follow-up study. Br. J. Cancer 2012, 106, 1171–1176. [Google Scholar] [CrossRef]
- Shields, C.L.; Ganguly, A.; Bianciotto, C.G.; Turaka, K.; Tavallali, A.; Shields, J.A. Prognosis of Uveal Melanoma in 500 Cases Using Genetic Testing of Fine-Needle Aspiration Biopsy Specimens. Ophthalmology 2011, 118, 396–401. [Google Scholar] [CrossRef]
- Tschentscher, F.; Prescher, G.; Horsman, D.E.; White, V.A.; Rieder, H.; Anastassiou, G.; Schilling, H.; Bornfeld, N.; Bartz-Schmidt, K.U.; Horsthemke, B.; et al. Partial deletions of the long and short arm of chromosome 3 point to two tumor suppressor genes in uveal melanoma. Cancer Res. 2001, 61, 3439–3442. [Google Scholar]
- Parrella, P.; Fazio, V.M.; Gallo, A.P.; Sidransky, D.; Merbs, S.L. Fine mapping of chromosome 3 in uveal melanoma: Identification of a minimal region of deletion on chromosomal arm 3p25.1–p25.2. Cancer Res. 2003, 63, 8507–8510. [Google Scholar] [PubMed]
- Cross, N.A.; Ganesh, A.; Parpia, M.; Murray, A.K.; Rennie, I.G.; Sisley, K. Multiple locations on chromosome 3 are the targets of specific deletions in uveal melanoma. Eye 2006, 20, 476–481. [Google Scholar] [CrossRef] [PubMed]
- van Gils, W.; Lodder, E.M.; Mensink, H.W.; Kiliç, E.; Naus, N.C.; Brüggenwirth, H.T.; van Ijcken, W.; Paridaens, D.; Luyten, G.P.; de Klein, A. Gene expression profiling in uveal melanoma: Two regions on 3p related to prognosis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4254–4262. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bajdak-Rusinek, K.; Kupnicka, P.; Kapczuk, P.; Siminska, D.; Chlubek, D.; Baranowska-Bosiacka, I. The Role of CXCL16 in the Pathogenesis of Cancer and Other Diseases. Int. J. Mol. Sci. 2021, 22, 3490. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.R.; Kalirai, H.; Sacco, J.J.; Azevedo, R.A.; Duckworth, A.; Slupsky, J.R.; Coulson, J.M.; Coupland, S.E. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J. Pathol. 2020, 250, 420–439. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, T.; Zhao, Q.; Xiao, B.; Guo, J. Using circular RNA hsa_circ_0000190 as a new biomarker in the diagnosis of gastric cancer. Clin. Chim. Acta 2017, 466, 167–171. [Google Scholar] [CrossRef]
- Mishra, S.; Bernal, C.; Silvano, M.; Anand, S.; Ruiz, I.A.A. The protein secretion modulator TMED9 drives CNIH4/TGFalpha/GLI signaling opposing TMED3-WNT-TCF to promote colon cancer metastases. Oncogene 2019, 38, 5817–5837. [Google Scholar] [CrossRef]
- Zou, Y.; Chen, B. Long non-coding RNA HCP5 in cancer. Clin. Chim. Acta 2021, 512, 33–39. [Google Scholar] [CrossRef]
- McCannel, T.A.; Burgess, B.L.; Rao, N.P.; Nelson, S.F.; Straatsma, B.R. Identification of Candidate Tumor Oncogenes by Integrative Molecular Analysis of Choroidal Melanoma Fine-Needle Aspiration Biopsy Specimens. Arch. Ophthalmol. 2010, 128, 1170–1177. [Google Scholar] [CrossRef]
- Berns, K.; Hijmans, E.M.; Mullenders, J.; Brummelkamp, T.R.; Velds, A.; Heimerkx, M.; Kerkovan, R.M.; Madiredjo, M.; Nijwamp, W.; Weigelt, B.; et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004, 428, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Lleonart, M.E.; Vidal, F.; Gallardo, D.; Diaz-Fuertes, M.; Rojo, F.; Cuatrecasas, M.; Lopez-Vicente, L.; Kondoh, H.; Blanco, C.; Carnero, A.; et al. New p53 related genes in human tumors: Significant downregulation in colon and lung carcinomas. Oncol. Rep. 2006, 16, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Tschentscher, F.; Hüsing, J.; Hölter, T.; Kruse, E.; Dresen, I.G.; Jöckel, K.-H.; Anastassiou, G.; Schilling, H.; Bornfeld, N.; Horsthemke, B.; et al. Tumor Classification Based on Gene Expression Profiling Shows That Uveal Melanomas with and without Monosomy 3 Represent Two Distinct Entities. Cancer Res. 2003, 63, 2578–2584. [Google Scholar] [PubMed]
- Cheng, D.; Fan, J.; Ma, Y.; Zhou, Y.; Qin, K.; Shi, M.; Yang, J. LncRNA SNHG7 promotes pancreatic cancer proliferation through ID4 by sponging miR-342-3p. Cell Biosci. 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, C.; Hu, J.; Pan, Y.; Shan, Y.; Liu, B.; Jia, L. Long non-coding RNA-SNHG7 acts as a target of miR-34a to increase GALNT7 level and regulate PI3K/Akt/mTOR pathway in colorectal cancer progression. J. Hematol. Oncol. 2018, 11, 89. [Google Scholar] [CrossRef]
- Xia, Q.; Li, J.; Yang, Z.; Zhang, D.; Tian, J.; Gu, B. Long non-coding RNA small nucleolar RNA host gene 7 expression level in prostate cancer tissues predicts the prognosis of patients with prostate cancer. Medicine 2020, 99, e18993. [Google Scholar] [CrossRef]
- Wu, X.; Yuan, Y.; Ma, R.; Xu, B.; Zhang, R. lncRNA SNHG7 affects malignant tumor behaviors through downregulation of EZH2 in uveal melanoma cell lines. Oncol. Lett. 2020, 19, 1505–1515. [Google Scholar] [CrossRef]
- Vardanyan, S.; Brosig, A.; Merz, H.; Ranjbar, M.; Kakkassery, V.; Grisanti, S.; Tura, A. Metastasis of Uveal Melanoma with Monosomy-3 Is Associated with a Less Glycogenetic Gene Expression Profile and the Dysregulation of Glycogen Storage. Cancers 2020, 12, 2101. [Google Scholar] [CrossRef]
- Cheli, Y.; Giuliano, S.; Fenouille, N.; Allegra, M.; Hofman, V.; Hofman, P.; Bahadoran, P.; Lacour, J.P.; Tartare-Deckert, S.; Bertolotto, C.; et al. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2012, 31, 2461–2470. [Google Scholar] [CrossRef]
- Feige, E.; Yokoyama, S.; Levy, C.; Khaled, M.; Igras, V.; Lin Richard, J.; Lee, S.; Widlund Hans, R.; Granter Scott, R.; Kung Andrew, L.; et al. Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc. Natl. Acad. Sci. USA 2011, 108, E924–E933. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Maat, W.; Ly, L.V.; Jordanova, E.S.; de Wolff-Rouendaal, D.; Schalij-Delfos, N.E.; Jager, M.J. Monosomy of Chromosome 3 and an Inflammatory Phenotype Occur Together in Uveal Melanoma. Investig. Opthalmol. Vis. Sci. 2008, 49, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Souri, Z.; Wierenga, A.P.A.; Kilic, E.; Brosens, E.; Bohringer, S.; Kroes, W.G.M.; Verdijk, R.M.; van der Velden, P.A.; Luyten, G.P.M.; Jager, M.J. MiRNAs Correlate with HLA Expression in Uveal Melanoma: Both Up- and Downregulation Are Related to Monosomy 3. Cancers 2021, 13, 4020. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.J.; de Wolff-Rouendaal, D.; Breebaart, A.C.; Ruiter, D.J. Expression of HLA antigens in paraffin sections of uveal melanomas. Doc. Ophthalmol. 1986, 30, 69–76. [Google Scholar] [CrossRef] [PubMed]
- de Waard-Siebinga, I.; Hilders, C.G.; Hansen, B.E.; van Delft, J.L.; Jager, M.J. HLA expression and tumor-infiltrating immune cells in uveal melanoma. Graefes Arch. Clin. Exp. Ophthalmol. 1996, 234, 34–42. [Google Scholar] [CrossRef]
- de Lange, M.J.; Nell, R.J.; Lalai, R.N.; Versluis, M.; Jordanova, E.S.; Luyten, G.P.M.; Jager, M.J.; van der Burg, S.H.; Zoutman, W.H.; van Hall, T.; et al. Digital PCR-Based T-cell Quantification-Assisted Deconvolution of the Microenvironment Reveals that Activated Macrophages Drive Tumor Inflammation in Uveal Melanoma. Mol. Cancer Res. 2018, 16, 1902–1911. [Google Scholar] [CrossRef]
- Taherzadeh, S.; Sharma, S.; Chhajlani, V.; Gantz, I.; Rajora, N.; Demitri, M.T.; Kelly, L.; Zhao, H.; Ichiyama, T.; Catania, A.; et al. alpha-MSH and its receptors in regulation of tumor necrosis factor-alpha production by human monocyte/macrophages. Am. J. Physiol. 1999, 276, R1289–R1294. [Google Scholar] [CrossRef]
- Star, R.A.; Rajora, N.; Huang, J.; Stock, R.C.; Catania, A.; Lipton, J.M. Evidence of autocrine modulation of macrophage nitric oxide synthase by alpha-melanocyte-stimulating hormone. Proc. Natl. Acad. Sci. USA 1995, 92, 8016–8020. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Becher, E.; Mahnke, K.; Hartmeyer, M.; Schwarz, T.; Scholzen, T.; Luger, T.A. Evidence for the differential expression of the functional alpha-melanocyte-stimulating hormone receptor MC-1 on human monocytes. J. Immunol. 1997, 158, 3378–3384. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Rodriguez-Garcia, A.; Perez-Losada, J.; Sagrera, A.; Read, A.P.; Sanchez-Garcia, I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum. Mol. Genet. 2002, 11, 3231–3236. [Google Scholar] [CrossRef]
- Wang, N.; Li, Y.; Zhou, J. miR-31 Functions as an Oncomir Which Promotes Epithelial-Mesenchymal Transition via Regulating BAP1 in Cervical Cancer. Biomed Res. Int. 2017, 2017, 6361420. [Google Scholar] [CrossRef] [PubMed]
- Versluis, M.; de Lange, M.J.; van Pelt, S.I.; Ruivenkamp, C.A.; Kroes, W.G.; Cao, J.; Jager, M.J.; Luyten, G.P.; van der Velden, P.A. Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PLoS ONE 2015, 10, e0116371. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, A.E.; Verdijk, R.M.; Brouwer, R.W.; van den Bosch, T.P.; van den Berg, M.M.; Vaarwater, J.; Kockx, C.E.; Paridaens, D.; Naus, N.C.; Nellist, M.; et al. Clinical significance of immunohistochemistry for detection of BAP1 mutations in uveal melanoma. Mod. Pathol. 2014, 27, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- van Essen, T.H.; van Pelt, S.I.; Versluis, M.; Bronkhorst, I.H.; van Duinen, S.G.; Marinkovic, M.; Kroes, W.G.; Ruivenkamp, C.A.; Shukla, S.; de Klein, A.; et al. Prognostic parameters in uveal melanoma and their association with BAP1 expression. Br. J. Ophthalmol. 2014, 98, 1738–1743. [Google Scholar] [CrossRef] [PubMed]
- van Essen, T.H.; van Pelt, S.I.; Bronkhorst, I.H.G.; Versluis, M.; Némati, F.; Laurent, C.; Luyten, G.P.M.; van Hall, T.; van den Elsen, P.J.; van der Velden, P.A.; et al. Upregulation of HLA Expression in Primary Uveal Melanoma by Infiltrating Leukocytes. PLoS ONE 2016, 11, e0164292. [Google Scholar] [CrossRef] [PubMed]
- Souri, Z.; Wierenga, A.P.A.; Mulder, A.; Jochemsen, A.G.; Jager, M.J. HLA Expression in Uveal Melanoma: An Indicator of Malignancy and a Modifiable Immunological Target. Cancers 2019, 11, 1132. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Median (Min-Max) | |||
|---|---|---|---|
| Feature | Nr | MITF Expression | p Value |
| Gender | |||
| Male | 33 | 10.44 (9.4–11.3) | 0.60 a |
| Female | 31 | 10.48 (9.6–11.6) | |
| Iris colour e | |||
| Light | 29 | 10.52 (9.6–11.4) | 0.28 a |
| Dark | 8 | 10.25 (10.0–11.1) | |
| Tumour pigmentation f | |||
| Light | 43 | 10.59 (9.4–12.0) | 0.003 a |
| Dark | 20 | 10.21 (9.6–10.8) | |
| Cell type | |||
| Spindle cell | 22 | 10.50 (9.6–11.5) | 0.76 a |
| Epithelioid-mixed cell | 42 | 10.41 (9.4–11.6) | |
| Ciliary body involvement | |||
| No | 39 | 10.48 (9.4–11.6) | 0.64 a |
| Yes | 25 | 10.38 (9.6–11.1) | |
| TNM stage | |||
| I-IIB | 36 | 10.50 (9.6–11.6) | 0.28 a |
| IIIA-IIIC | 26 | 10.35 (9.4–11.1) | |
| Chromosome 3 status | |||
| Disomy | 24 | 10.77 (10.1–11.6) | <0.001 a |
| Monosomy | 40 | 10.27 (9.4–11.0) | |
| Chromosome 8q status | |||
| Normal | 13 | 10.84 (9.7–11.4) | 0.02 b |
| Gain | 23 | 10.48 (9.6–11.6) | |
| Amplification | 27 | 10.32 (9.4–11.0) | |
| Chromosome 6p status | |||
| Normal | 43 | 10.35 (9.6–11.6) | 0.049 a |
| Gain | 21 | 10.53 (9.4–11.5) | |
| BAP1 expression (IHC) e | |||
| BAP1 positive | 25 | 10.53 (9.6–11.4) | 0.002 a |
| BAP1 negative | 31 | 10.35 (9.4–11.5) | |
| Age at enucleation | −0.063 c | 0.62 d | |
| Largest Basal Diameter | −0.168 c | 0.18 d | |
| Thickness | −0.122 c | 0.34 d |
| Mean (±SD) | Correlation Coeff. | Sig (2-Tailed) | |
|---|---|---|---|
| MC1R | 7.95 (±0.6) | −0.406 | 0.001 |
| MLANA | 13.73 (±0.4) | −0.259 | 0.038 |
| PMEL | 14.38 (±0.4) | −0.123 | 0.33 |
| TYR | 12.08 (±0.6) | 0.182 | 0.15 |
| TYRP1 | 13.66 (±0.9) | −0.379 | 0.002 |
| RAB 27a pr1 | 8.56 (±0.5) | −0.116 | 0.36 |
| RAB 27a pr3 | 7.95 (±0.5) | 0.332 | 0.007 |
| OA1 | 9.84 (±0.6) | −0.106 | 0.41 |
| Mean (±SD) | Correlation Coeff. | Sig (2-Tailed) | |
|---|---|---|---|
| CD3D | 7.13 (±1.1) | −0.249 | 0.048 |
| CD3E | 6.51 (±0.3) | −0.203 | 0.11 |
| CD4 | 6.66 (±0.3) | −0.237 | 0.06 |
| CD8A pr1 | 7.19 (±1.1) | −0.220 | 0.08 |
| CD8A pr3 | 7.26 (±1.3) | −0.315 | 0.011 |
| CD68 pr1 | 10.84 (±0.9) | −0.404 | 0.001 |
| CD68 pr2 | 9.45 (±0.9) | −0.359 | 0.004 |
| CD163 pr3 | 6.62 (±0.3) | −0.091 | 0.48 |
| FOXP3 | 6.50 (±0.1) | −0.057 | 0.65 |
| HLA-A pr1 | 11.36 (±1.0) | −0.363 | 0.003 |
| HLA-A pr2 | 13.84 (±0.8) | −0.429 | <0.001 |
| HLA-A pr4 | 10.71 (±1.4) | −0.379 | 0.002 |
| HLA-B | 11.33 (±1.7) | −0.358 | 0.004 |
| HLA-DRA pr1 | 10.49 (±1.4) | −0.153 | 0.23 |
| HLA-DRA pr2 | 11.34 (±1.5) | −0.228 | 0.07 |
| Mean (±SD) | Correlation Coeff. | Sig (2-Tailed) | |
|---|---|---|---|
| SNAI2 pr1 | 8.17 (±0.6) | 0.413 | 0.001 |
| SNAI2 pr2 | 9.49 (±0.7) | 0.278 | 0.026 |
| ZEB2 | 7.67 (±0.3) | 0.501 | <0.001 |
| TWIST1 | 6.60 (±0.2) | −0.263 | 0.036 |
| STAT 3 pr1 | 8.02 (±0.4) | −0.146 | 0.25 |
| STAT3 pr2 | 9.05 (±0.4) | −0.145 | 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelmi, M.C.; Verdijk, R.M.; Houtzagers, L.E.; van der Velden, P.A.; Kroes, W.G.M.; Luyten, G.P.M.; Vu, T.H.K.; Jager, M.J. Microphthalmia-Associated Transcription Factor: A Differentiation Marker in Uveal Melanoma. Int. J. Mol. Sci. 2023, 24, 8861. https://doi.org/10.3390/ijms24108861
Gelmi MC, Verdijk RM, Houtzagers LE, van der Velden PA, Kroes WGM, Luyten GPM, Vu THK, Jager MJ. Microphthalmia-Associated Transcription Factor: A Differentiation Marker in Uveal Melanoma. International Journal of Molecular Sciences. 2023; 24(10):8861. https://doi.org/10.3390/ijms24108861
Chicago/Turabian StyleGelmi, Maria Chiara, Robert M. Verdijk, Laurien E. Houtzagers, Pieter A. van der Velden, Wilma G. M. Kroes, Gregorius P. M. Luyten, T. H. Khanh Vu, and Martine J. Jager. 2023. "Microphthalmia-Associated Transcription Factor: A Differentiation Marker in Uveal Melanoma" International Journal of Molecular Sciences 24, no. 10: 8861. https://doi.org/10.3390/ijms24108861