1. Introduction
Monoclonal antibody drugs have been transformative for the treatment of many different diseases during the past decades, most notably in the areas of autoimmune disorders and oncology. Although the real breakthrough in neurodegenerative disorders is still yet to be demonstrated, the recent results from the pivotal Clarity AD Phase 3 study of lecanemab is encouraging [
1].
In parallel to the continued development and refinement of antibodies as drugs, research has also been focused on exploring non-antibody-based affinity protein as biopharmaceuticals [
2,
3]. Affibody molecules is a class of small (58 amino acids) affinity proteins with a three-helical structure that has been evaluated relatively extensively for in vivo diagnostics in oncology and as therapeutic drugs in both oncology and autoimmune diseases. The most advanced affibody molecule is currently tested in clinical phase III trials for treatment of different IL17A-driven autoimmune conditions and the results so far demonstrate excellent efficacy and safety [
4].
In a previous effort to develop affibody molecules for amyloid beta (Aβ), involved in the pathogenesis of Alzheimer’s disease (AD), phage display technology was used for selection of binders for the peptide. Screening of hits from the selection showed that the effort was successful, and analysis of the top candidate (denoted Z
Aβ3) revealed that the directed evolution had resulted in a new type of binder that was distinct from the original affibody protein [
5]. Although the genetic sequence was similar, the three-dimensional structure and mode of binding differed quite dramatically from other reported affibody molecules. Most notably, the binder was shown to fold as a disulphide-stabilized homodimer formed by an evolved cysteine at a specific position in each subunit. Moreover, enrichment of helix-destabilizing prolines and glycines in the
N-terminal region of the protein resulted in loss of the first helix in respective subunit [
6]. Upon binding, this region co-folded with the Aβ peptide forming a four-stranded beta sheet that stabilized the interaction. In the formed complex, the aggregation-prone parts of the Aβ peptide were buried in a tunnel-like cavity, and it was later demonstrated that this resulted in inhibition of aggregation [
5,
6,
7]. The Aβ-binder has been further engineered and a new variant with picomolar affinity (denoted Z
SYM73) has been evaluated with encouraging results in experimental therapy studies in AD animal models [
8,
9,
10].
Efforts have also been made to investigate whether the new type of dimeric affibody could be engineered for new specificities. Using error-prone PCR combined with phage display technology, several new binders against other aggregation-prone peptides, such as alpha synuclein, tau and islet amyloid polypeptide, where discovered, showing an almost identical structure and mode of binding as the parental amyloid-beta binder [
11,
12,
13]. Although the affinities were modest and several binders demonstrated a relatively high degree of cross-reactivity for different peptides, the studies showed that it was possible to engineer the specificity of this new class of affinity proteins.
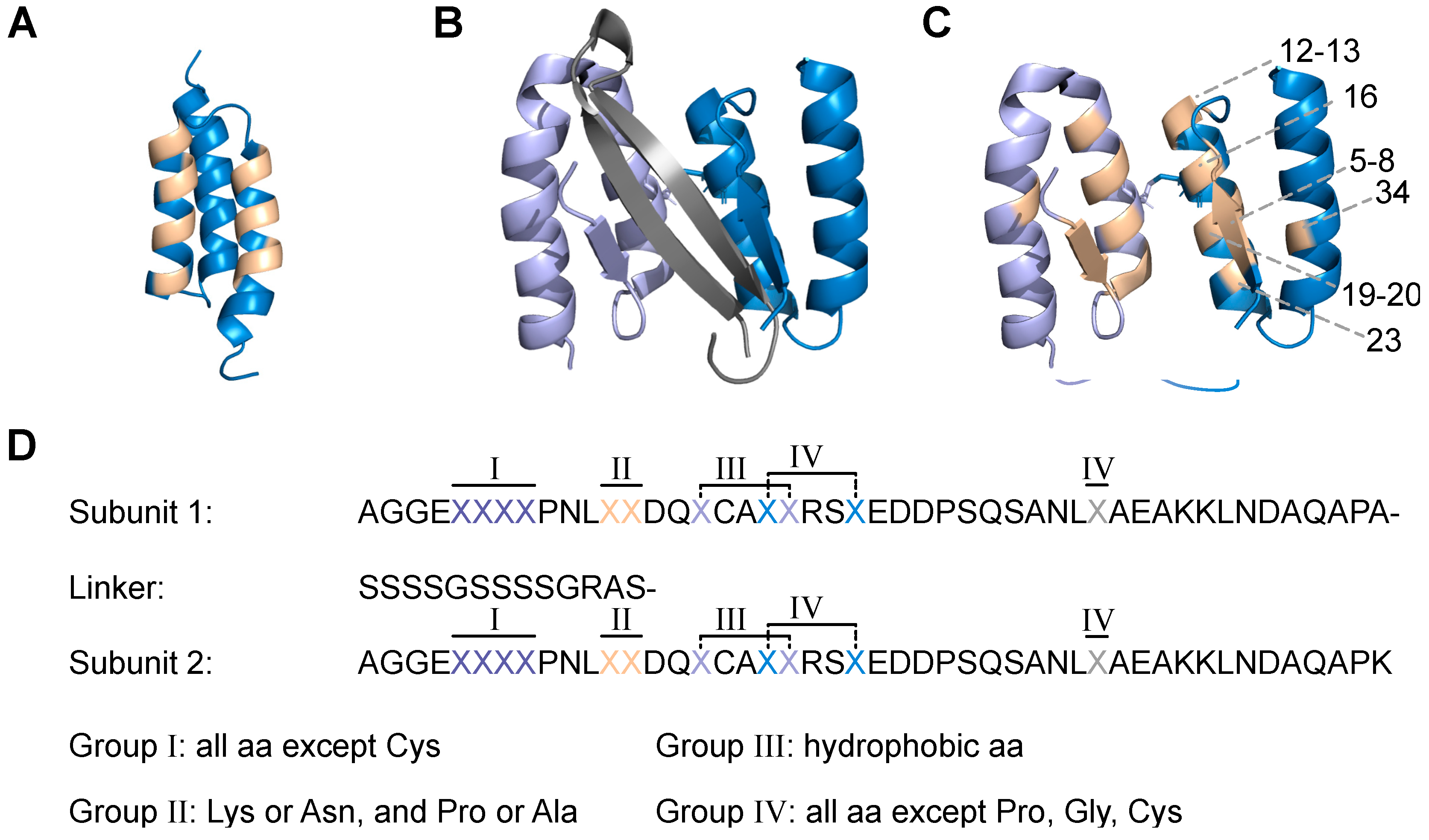
Encouraged by these studies, the aim here was to engineer the dimeric affibody into a scaffold protein that could be used as basis for generation of high-diversity libraries, followed by selection of new aggregation inhibitors. The scaffold was designed based on previously reported data [
10,
14,
15], including an
N-terminal truncation and a glycine/serine linker to form a head-to-tail dimeric fusion protein. From previously reported mutational studies and three-dimensional structures of similar dimeric affibody binders in complex with respective target peptides [
10,
15], eleven positions in each subunit were targeted for randomisation. Subcloning of the designed DNA library yielded a phage-displayed library with 5 × 10
9 diversity. To assess the quality of the library, a proof-of-principle selection against Aβ was performed, yielding high-affinity binders in the low nanomolar range. The variants with highest affinity were finally analysed with respect to their effect on Aβ aggregation and the results demonstrated complete inhibition of fibril formation at a 1:1 molar ratio. Due to their capacity of sequestering aggregation-prone peptides, we propose to call this new class of binders
Sequestrins.
3. Discussion
Neurodegenerative disorders are a huge burden for society and the burden is expected to grow with the increasing global life expectancy [
20]. A better understanding of the molecular mechanisms behind such diseases and finding suitable diagnostics and treatment strategies is thus essential.
In an effort to contribute with new molecular tools for studies and inhibition of peptide aggregation in neurodegenerative disorders, a new protein scaffold was designed, intended for generation of affinity reagents for peptides such as the amyloid beta (Aβ) peptide.
Based on data from previous research on related affibody-based peptide binders, a head-to-tail dimeric protein scaffold (denoted sequestrin) was designed and used for construction of a combinatorial protein library, containing around 5 × 10
9 variants. The sequestrin library was displayed on phage and used in biopanning against the Aβ peptide to assess the utility of the scaffold and the library design in respect of development of peptide-binding affinity reagents. The library design resulted in a theoretical diversity of 1.27 × 10
22 variants. The obtained phage library, containing 5 × 10
9 individual clones, is thus only covering a very small fraction of all possible combinations. However, the library was intentionally designed to include many randomized positions in the protein, given the limited amount of information on this new type of protein scaffold and mode of binding. The long-term aim is that selections, followed by sequencing and characterization of binders will result in knowledge that will guide the design of improved next-generation sequestrin libraries in the future. Moreover, the recent substantial improvement in computational methods for predicting protein folding and protein–protein interactions, such as neural network-based models [
21], paves the way for computer-aided library design based on deep-sequencing data on naïve, displayed and enriched library clones in the future.
A tendency of growth bias was observed during amplification of the phagemid library, which is likely due to the more complex protein structure compared to monomeric affibody molecules. Adjusting the amplification conditions during cultivation as described above was critical for ensuring a successful selection procedure.
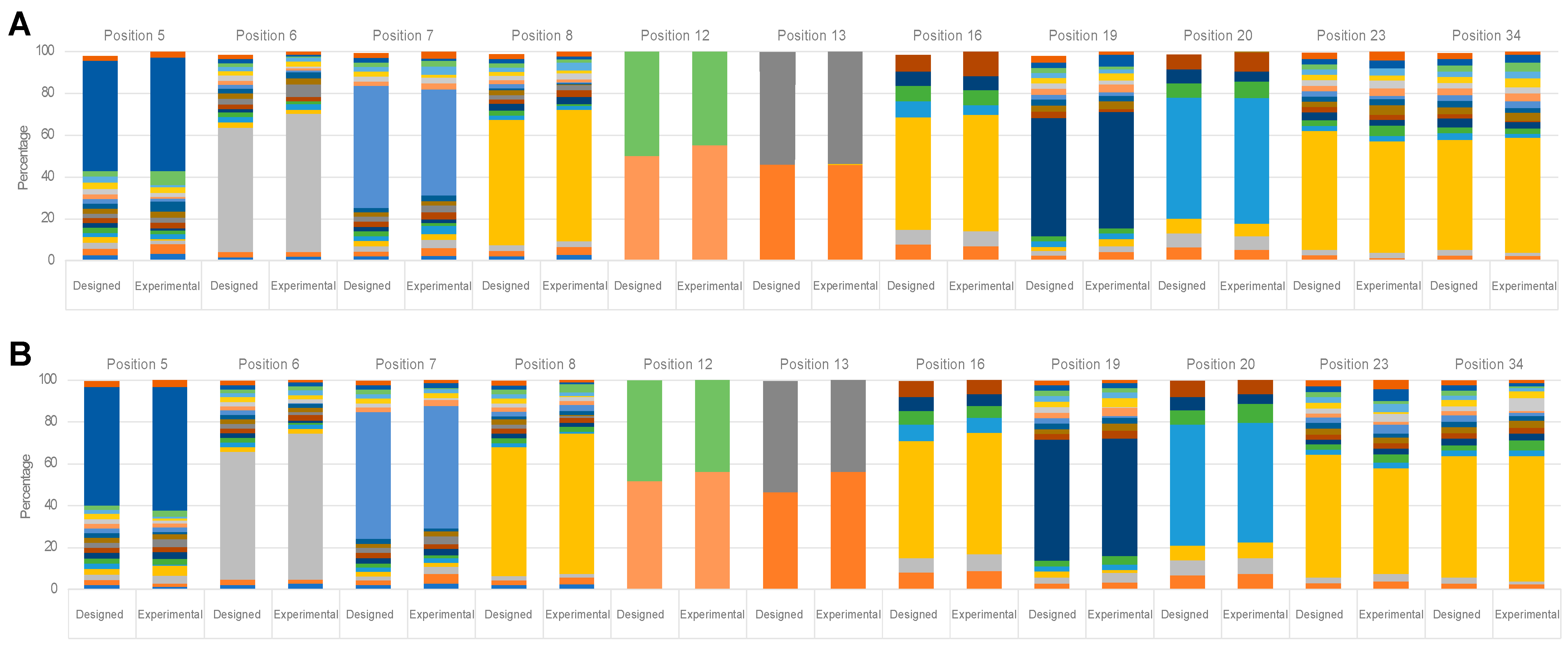
Sequencing of the naïve library revealed that the average number of mutations per sequestrin was around seven, which after selection had shifted to around four among the enriched peptide-binding variants. Future selections against other targets will show if this trend continuous and could hopefully be valuable for further developments of the sequestrin scaffold for protein engineering.
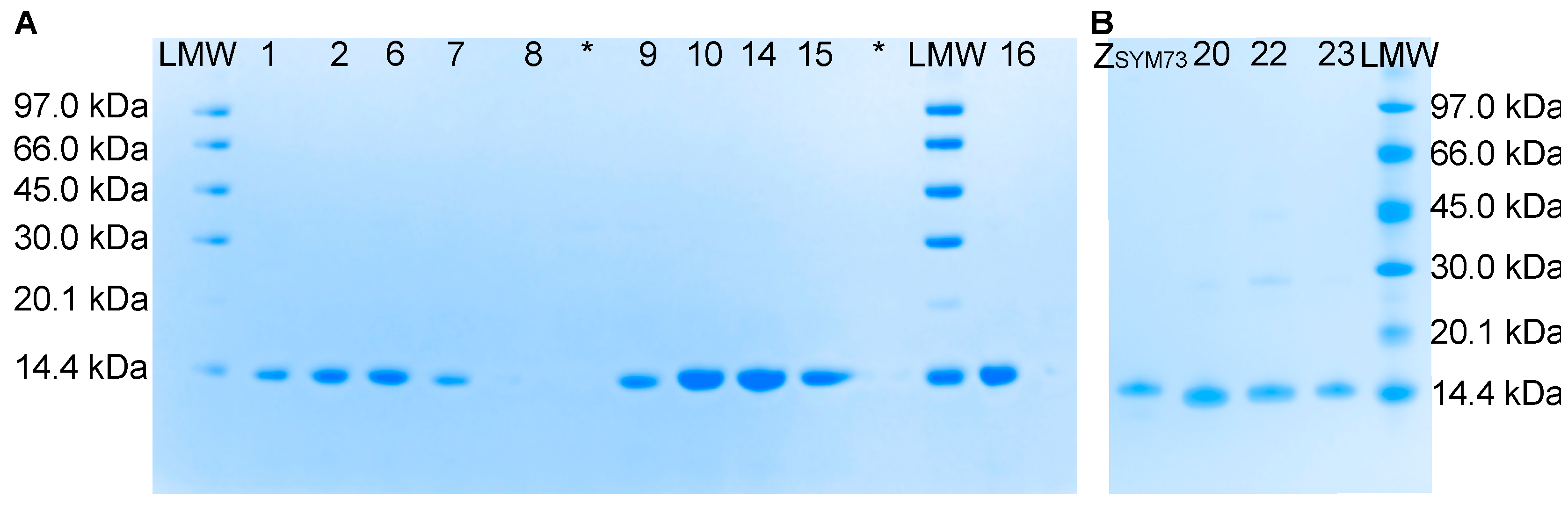
Based on the most reoccurring sequences and taking into account representation from the different selection cycles (3, 4, and 5), 13 clones were selected for characterisation. The thirteen sequestrins were produced, purified, and initially analysed in terms of secondary structure content, thermal stability, refolding and affinity.
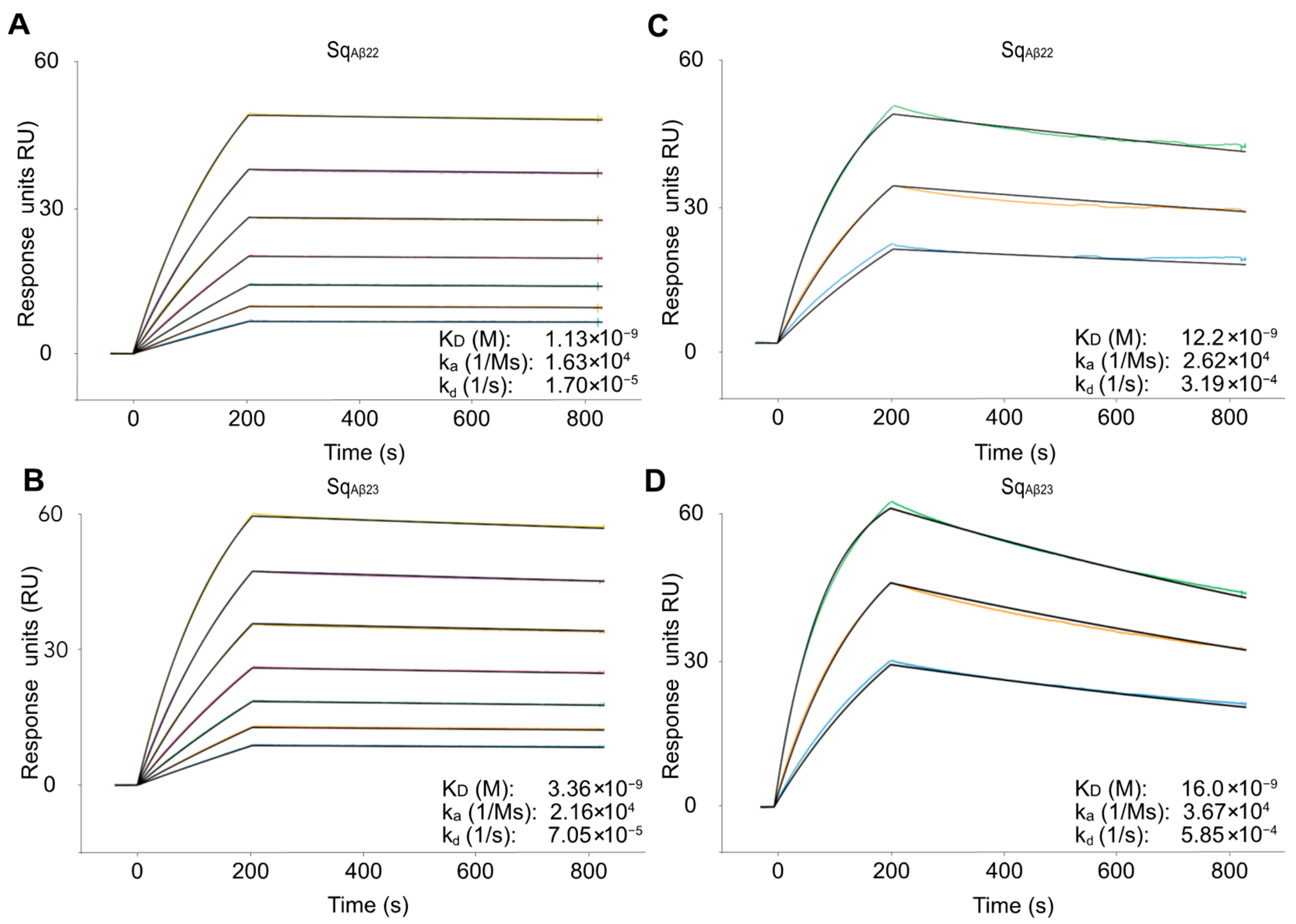
Eleven of the thirteen candidates showed high affinity for the amyloid-beta peptide with K
D in the 1–30 nM range, characterized by relatively slow kinetics for both the association and the dissociation. The variants with highest affinity (Sq
Aβ22 and Sq
Aβ23) originated from the fifth selection cycle, indicating that the increased stringency in the later cycles indeed enriched for high-affinity binding. As expected, analysis of the interaction at 37 °C demonstrated faster dissociation, corresponding to around 5- to 10-fold higher K
D. It should be noted that immobilizing the Aβ peptide on the chip probably results in an underestimation of the affinity. Previously reported SPR-based studies on structurally related dimeric affibody molecules for Aβ revealed a relatively large difference when the affinity was determined using SPR compared with analysis in solution [
10]. This is likely due to the structural rearrangements of both interactants upon binding [
7], which might be slower when the Aβ peptide is immobilized on the chip surface.
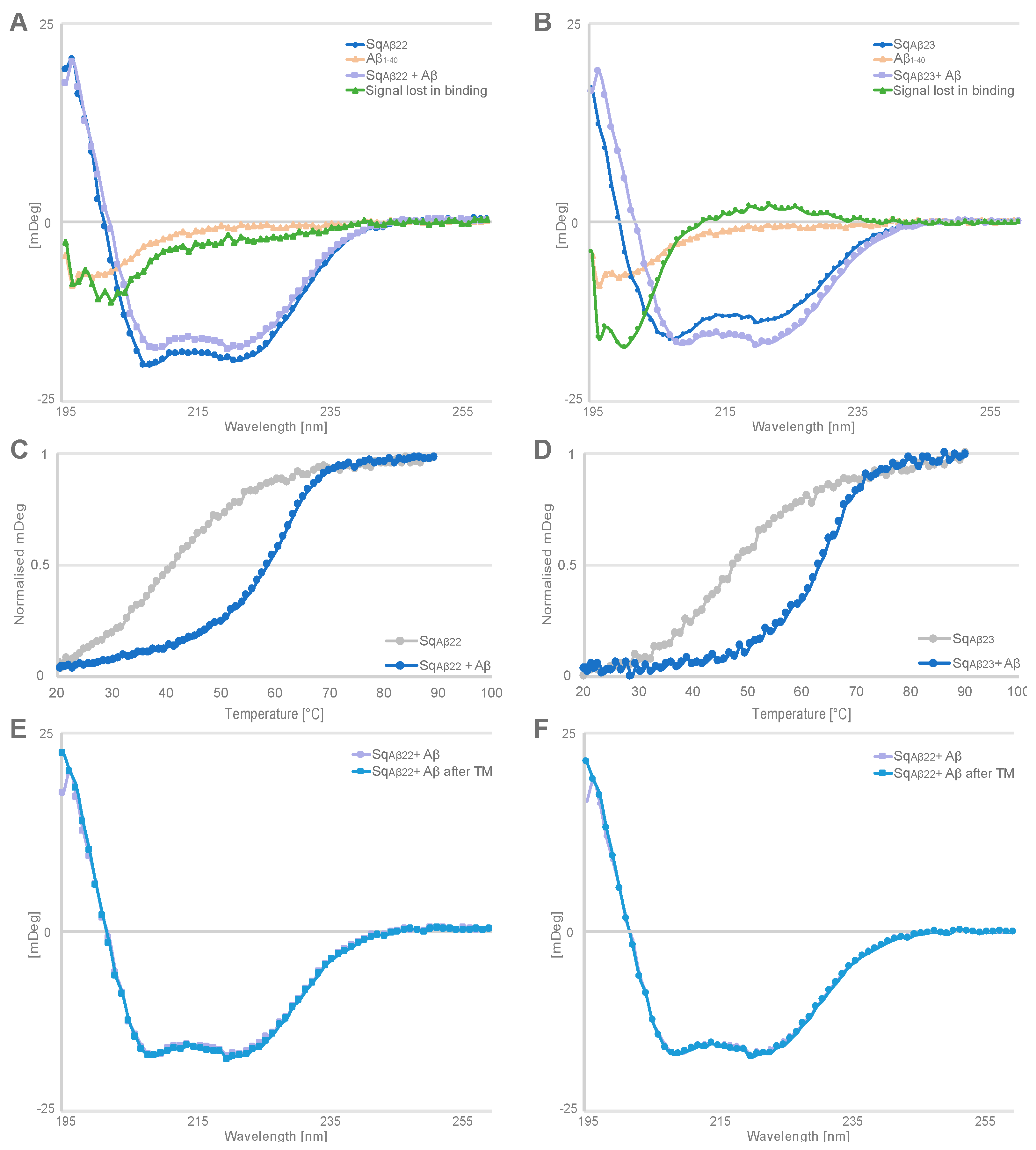
To get an indication whether similar structural rearrangement also occurs when the new sequestrins bind to the Aβ peptide, circular dichroism (CD) spectroscopy was used to detect potential changes in secondary structure content upon binding. The analysis showed that the Aβ peptide is unstructured in solution and the secondary structure content of the sequestrins is largely alpha helical. Co-incubation of Aβ peptide and respective sequestrin resulted in changes in the CD spectrum compared with the sum of CD spectra of the free interactants. Moreover, the thermal stability of the complex was increased 17–20 °C compared to the T
m for the free sequestrins. In future studies, determining the three-dimensional structure of the complex would confirm whether these results are indeed due to structure rearrangements in the sequestrins and the Aβ peptide, similar to what has been observed for dimeric peptide-binding affibody molecules [
7].
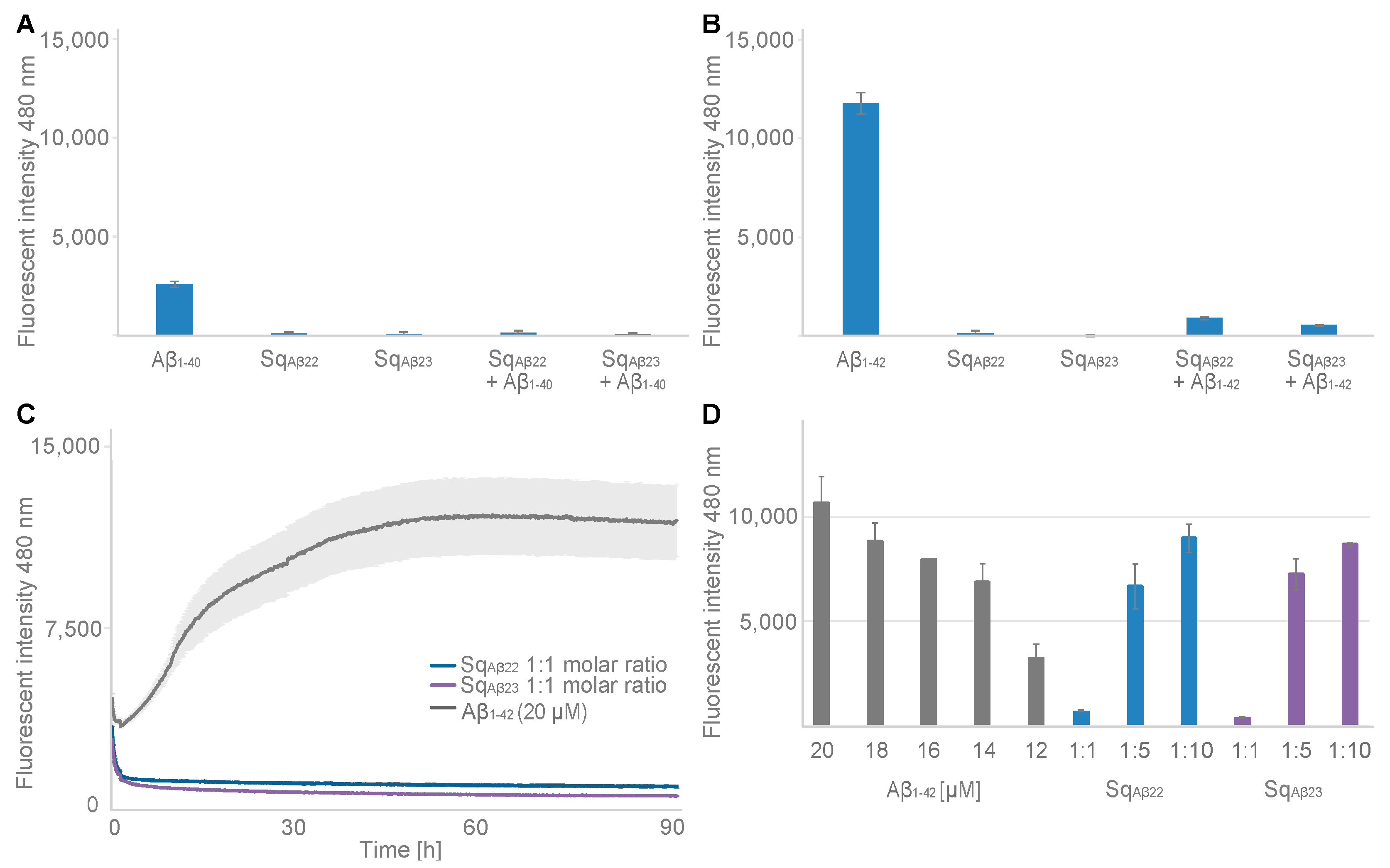
Finally, the effect from binding on the aggregation propensity of Aβ was assessed using a ThT fluorescence assay. The results showed that both sequestrins fully inhibited aggregation of Aβ when added at a 1:1 molar ratio, both for Aβ
1–40 and for the more aggregation-prone variant Aβ
1–42. Future studies on the effects of the new sequestrins on Aβ pathology in AD models is hence warranted. A challenge when targeting neurodegenerative diseases with biologics is the limited uptake across the blood-brain-barrier. Hence, genetic fusion of the sequestrins with a domain for transferrin receptor (TfR)-mediated transcytosis transportation [
22] would be interesting.
In summary, the results show that the design of the new sequestrin scaffold and library is suitable for selection of high-affinity peptide binders using phage display technology. Future selections against other aggregation-prone peptides, such as alpha synuclein, tau and TDP-43 will hopefully further validity the utility of this new class of affinity reagent.
4. Materials and Methods
4.1. Library Design and Molecular Cloning
Two randomized double stranded DNA oligos, encoding each of the two sequestrin (Sq) subunits, were purchased from TWIST Bioscience (San Francisco, CA, USA) for assembly into one library oligo by hybridization of overlapping bases in each subunit: 5′-GCGGGTGGCGAANNNNNNNNNNNNCCGAACTTANNNNNNGACCAANNN-TGTGCCNNNNNNCGTAGTNNNGAAGATGATCCTAGTCAAAGCGCTAACTTG-NNNGCAGAAGCTAAAAAGCTAAATGATGCTCAGGCGCCGGCGAGCAGCAGCAGCGGGAGCAGCAGCAGCGGGCGCGCGAGTGCGGGTGGCGAGNNNNNN-NNNNNNCCGAACTTANNNNNNGACCAANNNTGTGCCNNNNNNCGTAGTNNNGAGGATGACCCTAGTCAAAGCGCTAACTTGNNNGCAGAAGCTAAAAAGCT-AAATGATGCTCAGGCGCCGAAA -3′ (with randomized codons illustrated as NNN). The genes were flanked by BamHI and SalI restriction sites for subcloning in fusion to a gene for an albumin-binding domain (ABD; [
17]) into the pAffi1 phagemid [
5]. Each subunit of the library was amplified by polymerase chain reaction (PCR) in 12 cycles using Phusion DNA polymerase (New England Biolabs, Ipswich, MA, USA) and primers introducing 60 overlapping bases into the two oligo subunits for subsequent hybridization. The PCR products were purified using a PCR purification kit (Qiagen GmbH, Hilden, Germany). Equimolar amounts of the oligo subunits were hybridized into one long library gene of 321 bp, which was subsequently PCR-amplified in 10 cycles using Phusion DNA polymerase (New England Biolabs, Ipswich, MA, USA). PCR products of correct length were purified by preparative gel electrophoresis (2% agarose gel) followed by purification using a QIAquick gel purification kit (Qiagen GmbH, Nordrhein-Westfalen, Germany). Purified PCR products were digested by BamHI and SalI (New England Biolabs, Ipswich, MA, USA) enzymes. The modified pAffi1 vector was digested by the same enzymes and purified by preparative gel electrophoresis. Purified sequestrin fragments were ligated to the phagemid vector as
N-terminal fusion to a gene encoding an albumin-binding domain (ABD) [
17], using T4 DNA ligase (New England Biolabs, Ipswich, MA, USA) at a 1:6 molar ratio of vector to insert. Ligated vector was purified using a QIAquick PCR purification kit (Qiagen GmbH, Germany) before transformed into TG1 electrocompetent cells (Lucigen, Middleton, WI, USA).
Phage stocks were created by standard procedures in XL1Blue cells (Aglient, Santa Clara County, CA, USA). Briefly, superinfection was performed using 5-fold excess of M13K07 phage (New England Biolabs, Ipswich, MA, USA) and precipitation of phage particles was performed using polyethylene glycol (PEG)/NaCl to yield phage titres of approximately 1013 pfu/mL.
4.2. Library Validation
Superinfected bacterial colonies were individually screened for (1) library size by titration, (2) insert length by PCR amplification using DreamTaq DNA Polymerase (Thermo Scientific, Waltham, MA, USA) and gel electrophoreses, and (3) DNA sequence by Sanger sequencing (Microsynth SeqLab, Gottingen, Germany). Monoclonal phage ELISA was used to validate expression of recombinant protein on the phage surface by ABD binding to human serum albumin (HSA). Individual clones were cultivated in 96-deep well format in tryptic soy broth medium supplemented with yeast extract (TSBY) and 100 μg/mL carbenicillin at 30 °C, 250 rpm, ON. After ON, cultures were re-inoculated and incubated at 37 °C, 250 rpm until OD600 reached 0.5–0.8 AU. Superinfection with 0.3 × 106 pfu/clone M13K07 (New England Biolabs, Ipswich, MA, USA) at 37 °C without rotation for 30 min proceeded induction with 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG; Chemtronica, Stockholm, Sweden) and further cultivation ON at 37 °C, 250 rpm, under additional antibiotic pressure from 30 μg/mL kanamycin. Harvested phage supernatants were incubated for 1 h in 384-microwell plates (Nunc, PS, Low binding, Hi-Edge, clear) precoated with HSA [5 μg/mL] or bovine serum albumin (BSA) [1 w/v%]. The ELISA plate was blocked with 1 w/v% BSA prior to phage incubation to minimize background, and plates were washed in phosphate-buffered saline + 0.05% Tween-20 (PBST) before incubating with a mouse monoclonal M13 Bacteriophage Antibody (HRP) (Sino biological, Beijing, China) according to supplier’s instructions. Signal development was done with Pierce™ TMB Substrate Kit (Thermo Scientific, Waltham, MA, USA), as by instructions. After colorimetric development, the reaction was terminated by addition of 2 M H2SO4. Absorbance was measured at 450 nm using a CLARIOStar Plus plate reader (BMG Labtech, Ortenberg, Germany). DNA sequencing was performed by Sanger sequencing (Microsynth SeqLab, Göttingen, Germany), and sequences analysed with the Geneious software (version 11.2, Biomatters LTD, Auckland, New Zealand).
4.3. Selections of Sequestrins against Amyloid Beta
The sequestrin library phage stock (denoted Sqlib) was used in selections against C-terminally biotinylated Aβ1–40 peptide (AnaSpec, Fremont, CA, USA) in a total of five rounds with decreasing amount of soluble target antigen in each round (50 nM, 40 nM, 20 nM, 10 nM, 1 nM). The incubation temperature varied between the cycles (4 °C ON for cycle 1, 1 h RT for cycle 2–4, 1 h 45 °C for cycle 5) before antigen was captured by Dynabeads M-280 Streptavidin beads (Invitrogen, Waltham, MA, USA). All cycles were preceded with a negative selection round towards beads pre-blocked with 5 w/v% BSA. The stringency of PBSTB (0.1% Tween-20, 3 w/v% BSA) washes was increased with each selection round (2 × 1 min, 4 × 1 min, 5 × 3 min, 5 × 6 min, 4 × 6 min + 1 × 2 h + 1 × 6 min), where the last wash was done in PBS buffer. Phage eluates were obtained by incubation for 10 min in 0.1 M glycine-HCl, pH 3.0, followed by neutralization by Tris-HCl, 1 M, pH 8.0. Eluates were amplified by infection with 100× excess of XL1Blue E. coli cells, followed by plating on Aquare BioAssay Dish (Corning, Somerville, MA, USA) with blood agar base (BAB; Merck, Darmstadt, Germany) supplemented with 2% D-glucose, 100 μg/mL carbenicillin, and 10 μg/mL tetracycline and incubated at 37 °C ON. Bacterial colonies were recovered by addition of TSBY medium to the plates, followed by scraping off colonies from the plates and dissolving the cells in TSBY before continuing cultivation in a suspended format. The cultures containing infected bacteria with the eluted phage (>100× excess compared to eluate complexity) were reinoculated to OD600 = 0.2 AU, followed by superinfection with five times excess of M13K07 phage at an OD600 = 0.8 AU.
4.4. Phage ELISA
Amplified phage stock and individual clones after each selection round were cultured for polyclonal and monoclonal phage ELISA, respectively, as described above. All phage stocks were diluted to the same concentration before incubating with target antigen.
ELISA plates in 384-well format (Nunc, PS, Low binding, Hi-Edge, clear) were prepared with four wells per individual sample according to: (1) one well coated with 5 μg/mL HSA, (2) one well coated with 1 w/v% BSA, (3) one well pre-coated with 5 μg/mL streptavidin followed by 1 μg/mL biotinylated amyloid beta peptide (Aβ1–40; AnaSpec, USA) and (4) one well pre-coated with 5 μg/mL streptavidin followed by 1 w/v% BSA. Development and washes were performed as described above. The signal, representing background, from wells with BSA (for 1) or streptavidin-BSA (for 3) were subtracted. The signal from wells with Aβ1–40 was normalized with the signal from wells with HSA. DNA sequences were identified by Sanger sequencing (Microsynth SeqLab, Göttingen, Germany), and the sequences were analysed with the Geneious software (version 11.2, Biomatters LTD, Auckland, New Zealand).
4.5. Expression and Purification of Soluble Sequestrins
Sequences from the selections were chosen for further characterization. Sequestrin-encoding DNA was amplified from phagemids with primers designed for the In-Fusion HD cloning kit (Takara Bio Europe, Göteborg, Sweden), according to manufacturer’s recommendations into the pET-26b(+)-vector for periplasmic production with a C-terminal His6-tag. Sequence-verified clones (Sanger sequencing, Eurofins Genomics GmbH, Ebersberg, Germany) were transformed using heat shock to E. coli BL21(DE3) for recombinant protein expression. Cultures were inoculated to an OD600 of 0.1 AU and incubated in TSBY with 25 μg/mL kanamycin at 37 °C until OD600 reached 1.0 AU before inducing with 1 mM IPTG and thereafter incubated at 25 °C for 16 h. Cell pellets from harvest were dissolved in running buffer [47 mM Na2HPO4, 3 mM NaH2PO4, 300 mM NaCl, 15 mM imidazole, pH 7.4] before proceeding with purification. Cells were sonicated using a Vibra-Cell VCX 130 sonicator (Sonics, Newtown, CT, USA) and cell debris was removed by centrifugation and filtration. Cell lysate was loaded on an equilibrated HisPur Cobalt Resin (Thermo Scientific, Waltham, MA, USA) and washed with running buffer. The sample was eluted with running buffer supplemented with 150 mM imidazole. Fractions of eluate containing protein according to analysis with the Pierce™ BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA), performed according to manufacturer’s instructions, were pooled and buffer exchanged to PBS on PD 10 desalting columns (Cytiva, Marlborough, MA, USA). The samples were analysed with Sodium Dodecyl Sulfate–PolyAcrylamide Gel (SDS-PAGE) (NuPAGE Bis-Tris 4–12%, Invitrogen, Waltham, MA, USA). Molecular mass was analysed by electrospray ionization mass spectrometry on a Thermo Ultimate3000 Bruker Impact II system connected to a ProSwift RP-4H, 1 × 50 mm column (Thermo Fisher, Waltham, MA, USA) using a linear gradient elution with acetonitrile (3 to 95%), supplemented with 0.1% formic acid. The instrument was run with settings for electrospray ionization with a positive ion polarity, spanning the mass range 500 to 50,000 m/z.
4.6. Circular Dichroism Spectroscopy
The secondary structure content of the sequestrins was analysed using circular dichroism spectroscopy on a Chirascan system (Applied Photophysics, Leatherhead, UK) with a 1 mm High precision cell (110-1P-40 cuvettes, Hellma Analytics, Munich, Germany). Five wavelength scans were recorded and averaged between 195 nm and 260 nm at 20 °C on 0.2 mg/mL protein in PBS. Melting point was determined by using a temperature gradient of 1 °C per minute at 221 nm for five average readings. The refolding capability was assessed by repeating the spectral scan after the sample had been subjected to heat treatment and cooled down to 20 °C. The spectra from before and after heating was compared to assess refolding. Respective sequestrin and Aβ
1–40 (AnaSpec, USA) were co-incubated at equimolar concentrations of 15.7 μM and analysed by circular dichroism for analysis of changes in secondary structure content upon interaction. Secondary structure content was approximated by BeStSel algorithm [
19].
4.7. Surface Plasmon Resonance Assay for Analysis of Binding to Aβ1–40
Surface plasmon resonance (SPR) on a Biacore 8K instrument (Cytiva, Marlborough, MA, USA) was used to determine affinity and kinetics of sequestrins binding to Aβ
1–40. Series S SA sensor chips (Cytiva, Marlborough, MA, USA) were immobilized with 120 response units (RU) of biotinylated Aβ
1–40 (AnaSpec, Fremont, CA, USA) according to manufacturer’s instructions. PBST (0.05% Tween-20) was used as running buffer. All protein candidates were injected as a multi-cycle analysis in an 8-step 1:1.5 dilution series from 342 nM to 30 nM at 25 °C in duplicate. The dimeric affibody Ztaq
3638-(G
4S)
2-Ztaq
3638-ABD was used as negative control [
18]. Analyte was injected for 200 s and dissociation was recorded for 600 s at 30 μL/min. Surfaces were regenerated with 10 mM HCl for 35 s and let to stabilize for 30 s before next cycle. The results were evaluated using the Multi-cycle kinetics method—1:1 binding, using the Biacore Insight Evaluation Software (Version 2.0.15, Cytiva, Marlborough, MA, USA).
In a second analysis, Series S SA sensor chips were immobilized with 80 response units (RU) of biotinylated Aβ1–40, and SqAβ22, and SqAβ23 were injected as analyte in a 1:2 dilution series ranging from 300 nM to 75 nM at 37 °C in duplicate.
4.8. Aggregation Assay
Fibrillization inhibition assays were done for the Aβ1–40 (#AS-24236, AnaSpec, Fremont, CA, USA), and Aβ1–42 (#AS24224, AnaSpec, Fremont, CA, USA) using the sequestrins SqAβ22 and SqAβ23 as inhibitors for aggregation at decreasing molar ratios 1:1, 1:5, and 1:10 in relation to the 20 μM of Aβ. All proteins were thawed on ice and spun down at 10,000× g for 5 min at 4 °C to remove precipitates. Each sample was prepared in triplicate in a volume of 60 μL per reaction by adding PBS (Aβ1–40: 10 mM phosphate and 150 mM NaCl, pH 7.2; Aβ1–42: 10 mM phosphate and 150 mM NaCl, pH 8.0), 20 μM Thioflavin T (ThT) dye, and 20 μM sequestrin before vortexing for 3 s and kept on ice. When all samples were prepared the Aβ peptide was added at 20 μM concentration to all sequestrin samples, or as a standard at 12–20 μM, to the tubes and vortexed for 1 s. The sample was added to a 384-well plate (Nunc, PS, Low binding, Hi-Edge, clear) with surrounding wells filled with PBS, and sealed to minimize evaporation during analysis. The fluorescence intensity was immediately analysed in a CLARIOStar Plus (BMG Labtech, Ortenberg, Germany) with excitation at 440–10 nm and emission at 480–10 nm. The readings were done during 999 cycles with a cycle time of 325 s at 37 °C with 50 flashes with 0.1 s settling time per well, using 1500 gain adjustment. The plate was shaken in an orbital motion 500 rpm for 1 s before each measurement. Analysis was done in the Mars analysis software (version 3.0, BMG Labtech, Ortenberg, Germany) where replicates were averaged, and blank subtracted from well containing only PBS with ThT dye.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}