
Structure-Guided Engineering of a Family IV Cold-Adapted Esterase Expands Its Substrate Range

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification of Potential Substrates for EstN7
2.2. EstN7WT Is Restricted to Short Acyl Chain Length Substrates
2.3. Probing the Molecular Determinants of EstN7 Substrate Range by Structure Guided Engineering
3. Materials and Methods
3.1. Structural Analysis
3.2. Protein Engineering and Protein Production
3.3. Enzyme Activity and Kinetics
3.4. Data-Mining for New Valuable Biotransformations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prakash, D.; Nawani, N.; Prakash, M.; Bodas, M.; Mandal, A.; Khetmalas, M.; Kapadnis, B. Actinomycetes: A Repertory of Green Catalysts with a Potential Revenue Resource. BioMed. Res. Int. 2013, 2013, 264020. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Engineering a more sustainable world through catalysis and green chemistry. J. R. Soc. Interface 2016, 13, 20160087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Si, T.; Zhao, H. Biocatalyst development by directed evolution. Bioresour. Technol. 2012, 115, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, P.J. The importance of Green Chemistry in Process Research and Development. Chem. Soc. Rev. 2011, 41, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, P.; Eck, J. Metagenomics and industrial applications. Nat. Rev. Genet. 2005, 3, 510–516. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. Microbial carboxyl esterases: Classification, properties and application in biocatalysis. FEMS Microbiol. Rev. 2002, 26, 73–81. [Google Scholar] [CrossRef]
- Arpigny, J.L.; Jaeger, K.E. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 1999, 343, 177–183. [Google Scholar] [CrossRef]
- Chandra, P.; Enespa; Singh, R.; Arora, P.K. Microbial lipases and their industrial applications: A comprehensive review. Microb. Cell Fact. 2020, 19, 169. [Google Scholar] [CrossRef]
- Sarmiento, F.; Peralta, R.; Blamey, J.M. Cold and Hot Extremozymes: Industrial Relevance and Current Trends. Front. Bioeng. Biotechnol. 2015, 3, 148. [Google Scholar] [CrossRef] [Green Version]
- Al-Ghanayem, A.A.; Joseph, B. Current prospective in using cold-active enzymes as eco-friendly detergent additive. Appl. Microbiol. Biotechnol. 2020, 104, 2871–2882. [Google Scholar] [CrossRef]
- Al-Maqtari, Q.A.; Waleed, A.-A.; Mahdi, A.A. Cold-active enzymes and their applications in industrial fields—A review. Int. J. Res. Agric. Sci. 2019, 6, 2348–3997. [Google Scholar]
- Noby, N.; Hussein, A.; Saeed, H.; Embaby, A.M. Recombinant cold—Adapted halotolerant, organic solvent-stable esterase (estHIJ) from Bacillus halodurans. Anal. Biochem. 2019, 591, 113554. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Ramteke, P.W.; Thomas, G. Cold active microbial lipases: Some hot issues and recent developments. Biotechnol. Adv. 2008, 26, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Kuddus, M. Cold-active microbial enzymes. Biochem. Physiol. 2015, 4, e132. [Google Scholar] [CrossRef]
- Elend, C.; Schmeisser, C.; Hoebenreich, H.; Steele, H.; Streit, W. Isolation and characterization of a metagenome-derived and cold-active lipase with high stereospecificity for (R)-ibuprofen esters. J. Biotechnol. 2007, 130, 370–377. [Google Scholar] [CrossRef]
- Rotticci, D.; Ottosson, J.; Norin, T.; Hult, K. Candida antarctica Lipase BA Tool for the Preparation of Optically Active Alcohols, Enzymes in Nonaqueous Solvents; Springer: Berlin/Heidelberg, Germany, 2001; pp. 261–276. [Google Scholar]
- Maharana, A.; Ray, P. A novel cold-active lipase from psychrotolerant Pseudomonas sp. AKM-L5 showed organic solvent resistant and suitable for detergent formulation. J. Mol. Catal. B Enzym. 2015, 120, 173–178. [Google Scholar] [CrossRef]
- Santiago, M.; Ramírez-Sarmiento, C.A.; Zamora, R.A.; Parra, L.P. Discovery, Molecular Mechanisms, and Industrial Applications of Cold-Active Enzymes. Front. Microbiol. 2016, 7, 1408. [Google Scholar] [CrossRef]
- Jemli, S.; Ayadi-Zouari, D.; Ben Hlima, H.; Bejar, S. Biocatalysts: Application and engineering for industrial purposes. Crit. Rev. Biotechnol. 2014, 36, 246–258. [Google Scholar] [CrossRef]
- Noby, N.; Auhim, H.S.; Winter, S.; Worthy†, H.L.; Embaby, A.M.; Saeed, H.; Hussein, A.; Pudney, C.R.; Rizkallah, P.J.; Wells, S.A.; et al. Structure and in silico simulations of a cold-active esterase reveals its prime cold-adaptation mechanism. Open Biol. 2021, 11, 210182. [Google Scholar] [CrossRef]
- Bassegoda, A.; Fillat, A.; Pastor, F.I.J.; Diaz, P. Special Rhodococcus sp. CR-53 esterase Est4 contains a GGG(A)X-oxyanion hole conferring activity for the kinetic resolution of tertiary alcohols. Appl. Microbiol. Biotechnol. 2013, 97, 8559–8568. [Google Scholar] [CrossRef]
- Noby, N.; Saeed, H.; Embaby, A.; Pavlidis, I.V.; Hussein, A. Cloning, expression and characterization of cold active esterase (EstN7) from Bacillus cohnii strain N1: A novel member of family IV. Int. J. Biol. Macromol. 2018, 120, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Bornscheuer, U.T. Activity of Lipases and Esterases towards Tertiary Alcohols: Insights into Structure–Function Relationships†. Angew. Chem. Int. Ed. 2002, 41, 3211–3213. [Google Scholar] [CrossRef]
- Zhu, X.; Larsen, N.A.; Basran, A.; Bruce, N.C.; Wilson, I.A. Observation of an Arsenic Adduct in an Acetyl Esterase Crystal Structure. J. Biol. Chem. 2003, 278, 2008–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, Y.; Ezaki, S.; Atomi, H.; Imanaka, T. Extremely Stable and Versatile Carboxylesterase from a Hyperthermophilic Archaeon. Appl. Environ. Microbiol. 2002, 68, 3925–3931. [Google Scholar] [CrossRef] [Green Version]
- Tutino, M.L.; di Prisco, G.; Marino, G.; de Pascale, D. Cold-adapted esterases and lipases: From fundamentals to application. Protein. Pept. Lett. 2009, 16, 1172–1180. [Google Scholar]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Genet. 2003, 1, 200–208. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Ribeiro, A.J.M.; Borkakoti, N.; Thornton, J.M. Transform-MinER: Transforming molecules in enzyme reactions. Bioinformatics 2018, 34, 3597–3599. [Google Scholar] [CrossRef]
- Chen, T.; Gong, T.; Zhao, T.; Liu, X.; Fu, Y.; Zhang, Z.; Gong, T. Paclitaxel loaded phospholipid-based gel as a drug delivery system for local treatment of glioma. Int. J. Pharm. 2017, 528, 127–132. [Google Scholar] [CrossRef]
- Palm, G.J.; Fernández-Álvaro, E.; Bogdanović, X.; Bartsch, S.; Sczodrok, J.; Singh, R.K.; Böttcher, D.; Atomi, H.; Bornscheuer, U.T.; Hinrichs, W. The crystal structure of an esterase from the hyperthermophilic microorganism Pyrobaculum calidifontis VA1 explains its enantioselectivity. Appl. Microbiol. Biotechnol. 2011, 91, 1061–1072. [Google Scholar] [CrossRef]
- McKary, M.G.; Abendroth, J.; Edwards, T.E.; Johnson, R.J. Structural Basis for the Strict Substrate Selectivity of the Mycobacterial Hydrolase LipW. Biochemistry 2016, 55, 7099–7111. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.K.; Radivojac, P.; Obradovic, Z.; Dunker, A.K.; Zhu, G. Improved amino acid flexibility parameters. Protein Sci. 2003, 12, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, H.; Yan, Q.; Yang, S.; Duan, X.; Jiang, Z. Biochemical Characterization of a First Fungal Esterase from Rhizomucor miehei Showing High Efficiency of Ester Synthesis. PLoS ONE 2013, 8, e77856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugihara, A.; Shimada, Y.; Nomura, A.; Terai, T.; Imayasu, M.; Nagai, Y.; Nagao, T.; Watanabe, Y.; Tominaga, Y. Purification and characterization of a novel cholesterol esterase from Pseudomonas aeruginosa, with its application to cleaning lipid-stained contact lenses. Biosci. Biotechnol. Biochem. 2002, 66, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Komiya, D.; Hori, A.; Ishida, T.; Igarashi, K.; Samejima, M.; Koseki, T.; Fushinobu, S. Crystal Structure and Substrate Specificity Modification of Acetyl Xylan Esterase from Aspergillus luchuensis. Appl. Environ. Microbiol. 2017, 83, e01251-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhl, P.; Doderer, K.; Hollmann, F.; Thum, O.; Pleiss, J. Engineering of Candida antarctica lipase B for hydrolysis of bulky carboxylic acid esters. J. Biotechnol. 2010, 150, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Santarossa, G.; Lafranconi, P.G.; Alquati, C.; DeGioia, L.; Alberghina, L.; Fantucci, P.; Lotti, M. Mutations in the “lid” region affect chain length specificity and thermostability of a Pseudomonas fragi lipase. FEBS Lett. 2005, 579, 2383–2386. [Google Scholar] [CrossRef] [Green Version]
- Holmquist, M. Insights into the molecular basis for fatty acyl specificities of lipases from Geotrichum candidum and Candida rugosa. Chem. Phys. Lipids 1998, 93, 57–65. [Google Scholar] [CrossRef]
- Peisajovich, S.G.; Tawfik, D.S. Protein engineers turned evolutionists. Nat. Methods 2007, 4, 991–994. [Google Scholar] [CrossRef]
- Bershtein, S.; Segal, M.; Bekerman, R.; Tokuriki, N.; Tawfik, D.S. Robustness–epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 2006, 444, 929–932. [Google Scholar] [CrossRef]
- Sideraki, V.; Huang, W.; Palzkill, T.; Gilbert, H.F. A secondary drug resistance mutation of TEM-1 beta -lactamase that suppresses misfolding and aggregation. Proc. Natl. Acad. Sci. USA 2000, 98, 283–288. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Ribeiro, A.J.M.; Borkakoti, N.; Thornton, J.M. Exploring Chemical Biosynthetic Design Space with Transform-MinER. ACS Synth. Biol. 2019, 8, 2494–2506. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Cumming, G.; Fidler, F.; Vaux, L.D. Error bars in experimental biology. J. Cell Biol. 2007, 177, 7–11. [Google Scholar] [CrossRef] [Green Version]





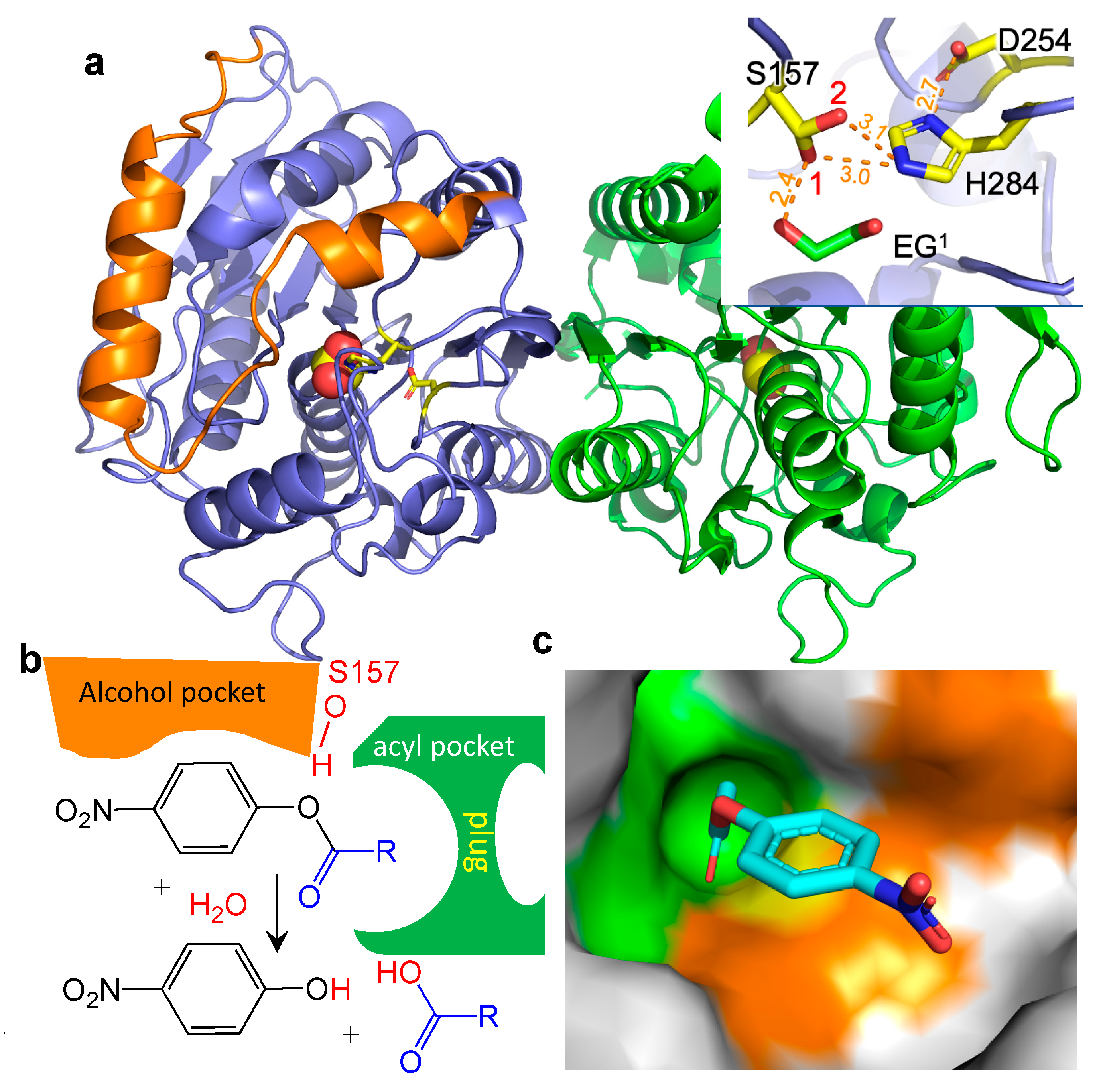{kind=link}
{kind=link}
{kind=link}
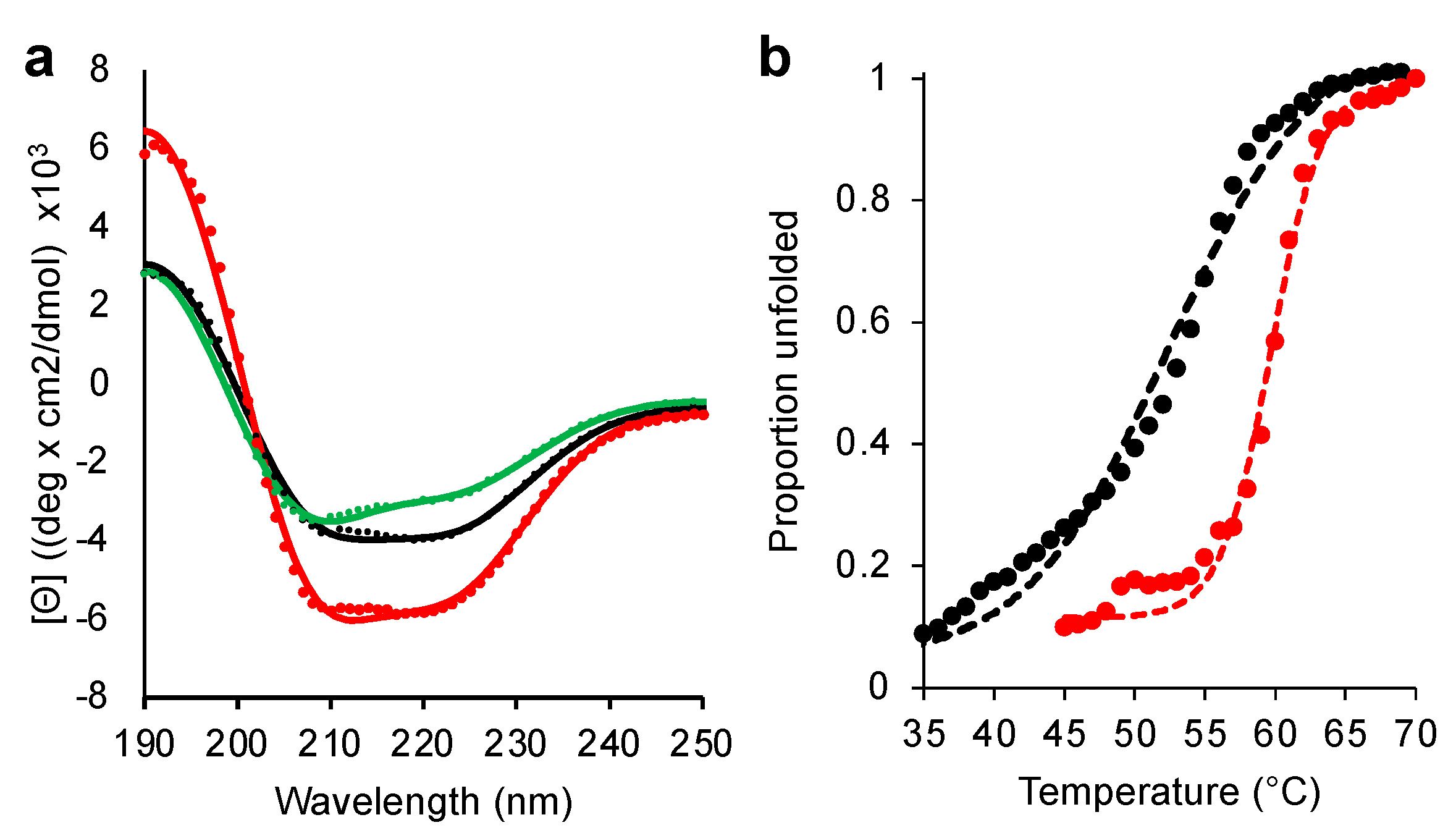{kind=link}
| EstN7 Variant | p-NF Substrate | KM (mM) | kcat (s−1) | Catalytic Efficiency (s−1 M−1) | Fold Change a |
|---|---|---|---|---|---|
| WT | C2 | 0.05 ± 0.007 | 760 ± 3.5 | 1.52 × 107 | 1 |
| C4 | 0.057 ± 0.002 | 0.32 ± 0.005 | 5614 | 1 | |
| C6 | - | - | - | ||
| C8 | - | - | - | ||
| N211A | C2 | 0.75 ± 0.001 | 280.5 ± 0.07 | 3.74 × 105 | 0.024 |
| C4 | 2 ± 0.2 | 15 ± 0.5 | 7500 | 1.33 | |
| C6 | 0.37 ± 0.05 | 2.3 ± 0.2 | 6283 | - | |
| C8 | - | - | - | - | |
| W206A | C2 | 1.3 ± 0.14 | 1.5 ± 0.07 | 1192 | 7.8 × 10−5 |
| C4 | 1.2 ± 0.55 | 8 ± 1.0 | 6666 | 1.2 | |
| C6 | 1.1 ± 0.07 | 3.8 ± 0.07 | 3347 | - | |
| C8 | - | - | - | ||
| DM (M187A/N211A) | C2 | 1.1 ± 0.07 | 361 ± 0.14 | 3.28 × 105 | 0.021 |
| C4 | 0.37 ± 0.08 | 17.55 ± 2.1 | 4.68 × 104 | 8.0 | |
| C6 | 1.2 ± 0.01 | 24.5 ± 0.7 | 2.04 × 104 | - | |
| C8 | 0.95 ± 0.02 | 0.45 ± 0.02 | 473 | - | |
| TM (M187A/W206A/N211A) | C2 | 2 ± 0.3 | 0.07 ± 0.01 | 35 | 2.31 × 10−6 |
| C4 | 2.1 ± 0.4 | 1 ± 0.11 | 467 | 0.08 | |
| C6 | 0.59 ± 0.03 | 1.45 ± 0.02 | 2457 | - | |
| C8 | 2 ± 0.03 | 0.85 ± 0.007 | 425 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noby, N.; Johnson, R.L.; Tyzack, J.D.; Embaby, A.M.; Saeed, H.; Hussein, A.; Khattab, S.N.; Rizkallah, P.J.; Jones, D.D. Structure-Guided Engineering of a Family IV Cold-Adapted Esterase Expands Its Substrate Range. Int. J. Mol. Sci. 2022, 23, 4703. https://doi.org/10.3390/ijms23094703
Noby N, Johnson RL, Tyzack JD, Embaby AM, Saeed H, Hussein A, Khattab SN, Rizkallah PJ, Jones DD. Structure-Guided Engineering of a Family IV Cold-Adapted Esterase Expands Its Substrate Range. International Journal of Molecular Sciences. 2022; 23(9):4703. https://doi.org/10.3390/ijms23094703
Chicago/Turabian StyleNoby, Nehad, Rachel L. Johnson, Jonathan D. Tyzack, Amira M. Embaby, Hesham Saeed, Ahmed Hussein, Sherine N. Khattab, Pierre J. Rizkallah, and D. Dafydd Jones. 2022. "Structure-Guided Engineering of a Family IV Cold-Adapted Esterase Expands Its Substrate Range" International Journal of Molecular Sciences 23, no. 9: 4703. https://doi.org/10.3390/ijms23094703