
Urolithin A Inactivation of TLR3/TRIF Signaling to Block the NF-κB/STAT1 Axis Reduces Inflammation and Enhances Antioxidant Defense in Poly(I:C)-Induced RAW264.7 Cells

 , ,
, ,  , and
, and 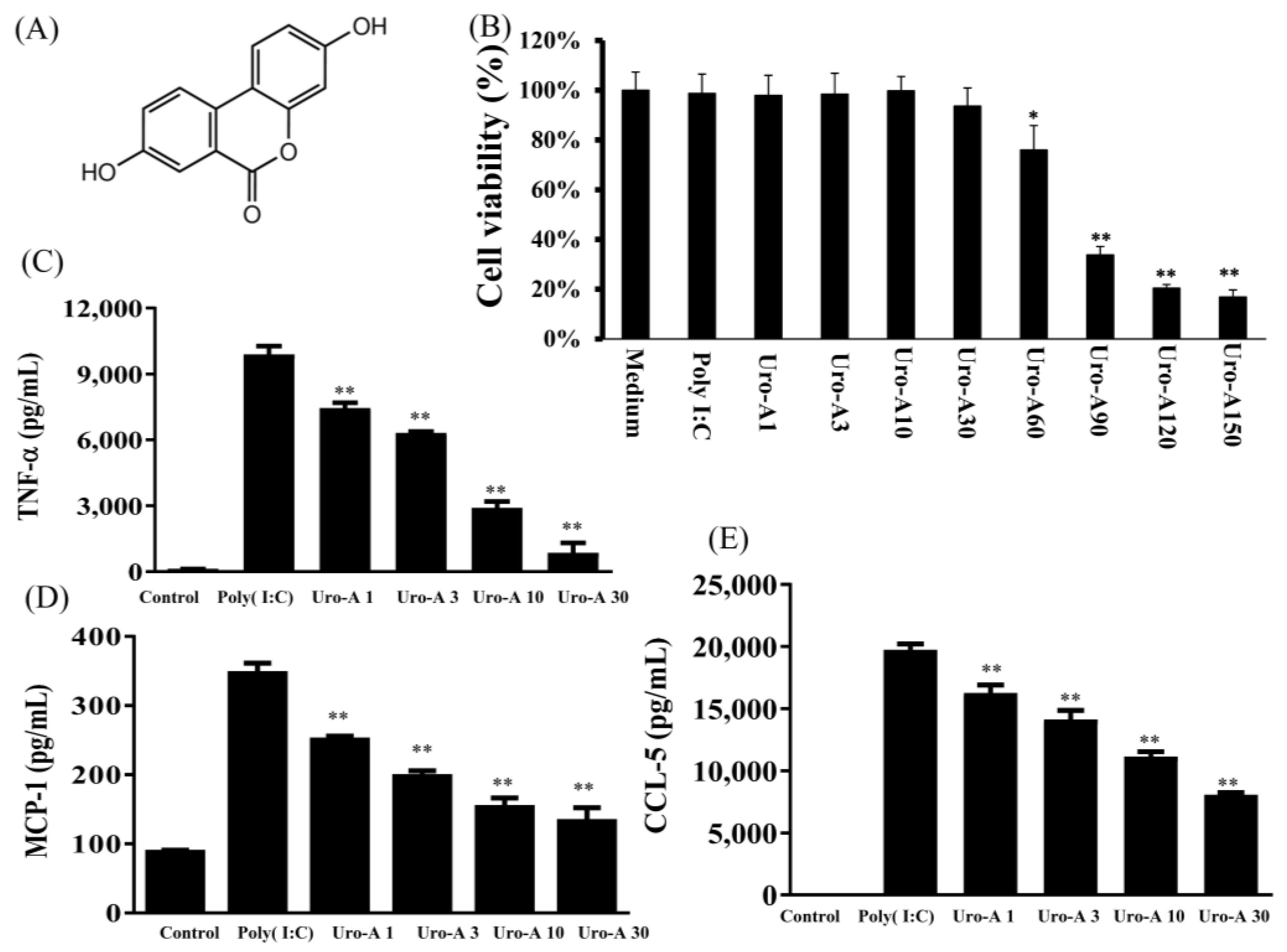{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Urolithin A Inhibited Inflammatory Cytokines and Inactived TLR3 Pathway Protein Expression in Poly(I:C)-Stimulated RAW264.7 Cells
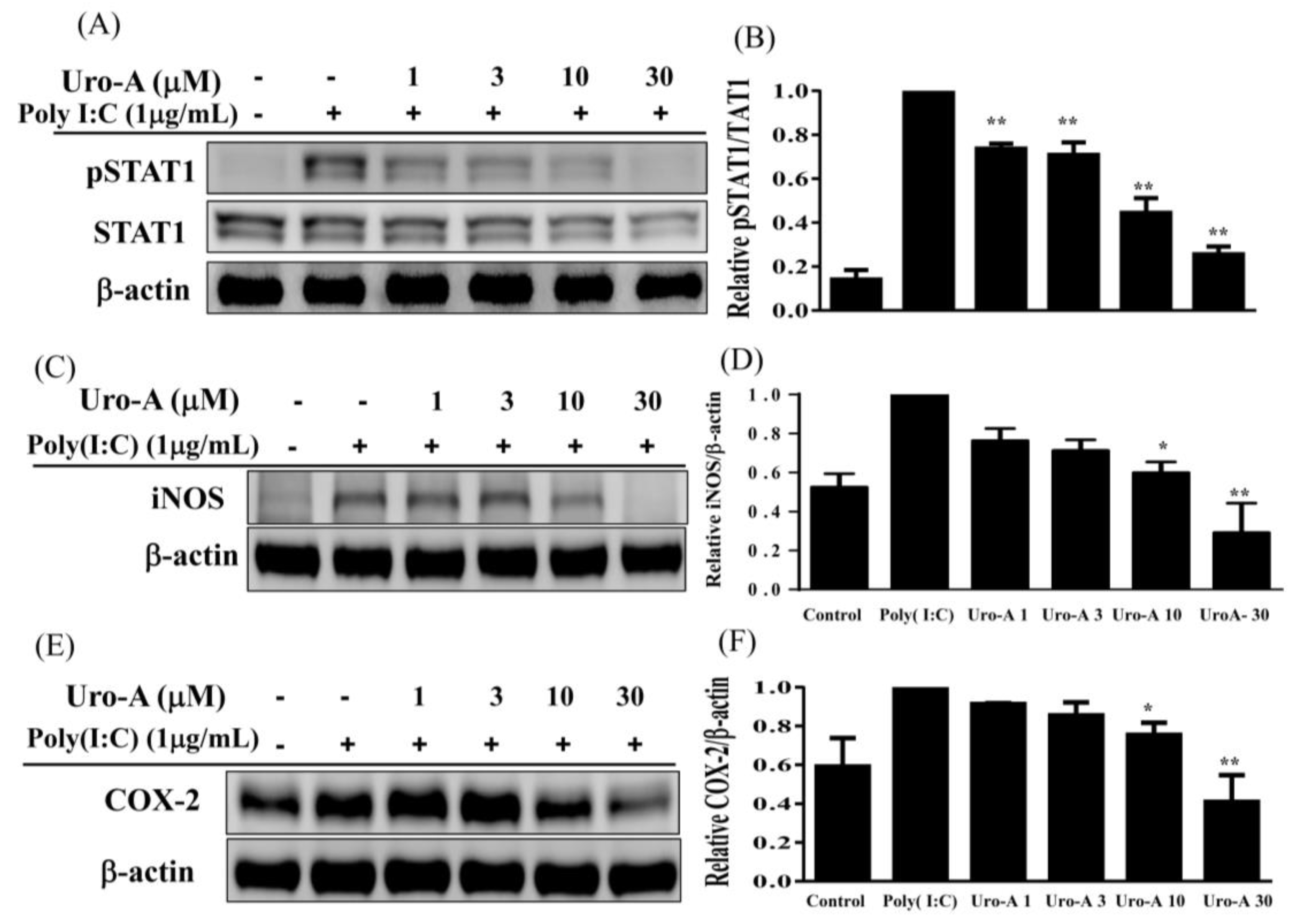
2.2. Urolithin A Blocked the NF-κB/STAT1 Pathway to Inhibit the Expression of Inflammatory Mediators in Poly(I:C)-Induced RAW264.7 Cells
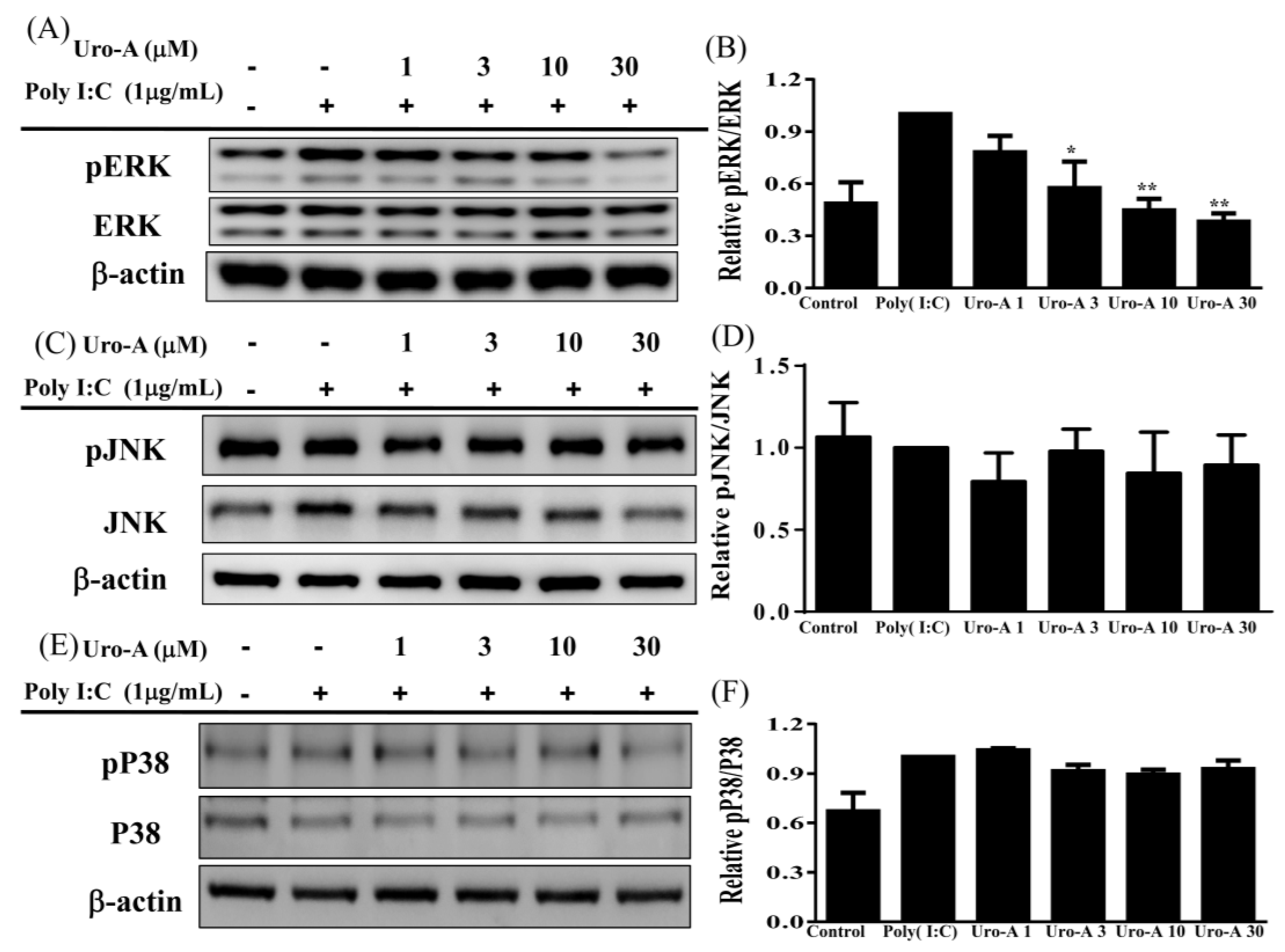
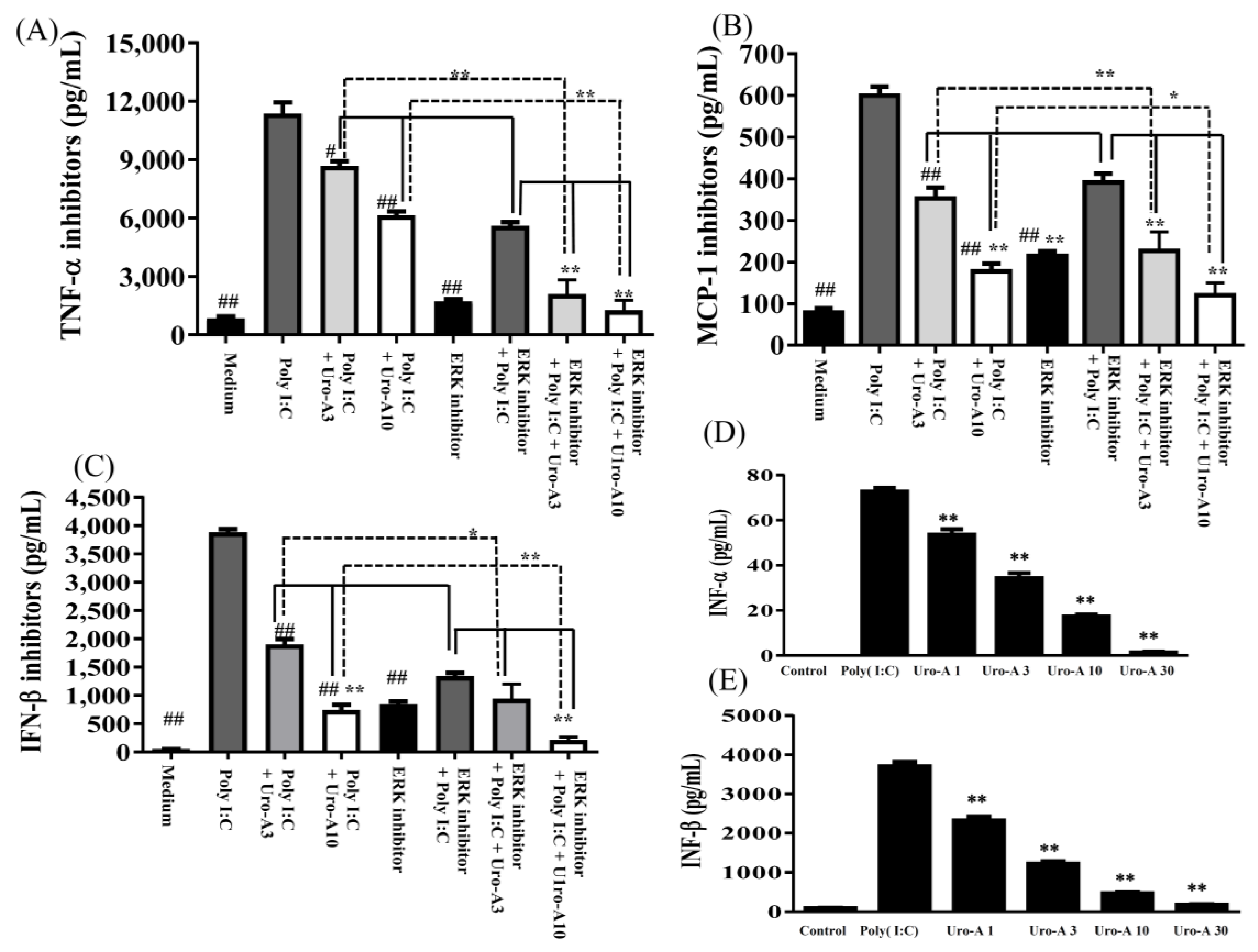
2.3. Urolithin A Suppressed the ERK/MAPK Pathway and the ERK Inhibitor Decreased Cytokine Secretion in Poly(I:C)-Stimulated RAW264.7 Cells
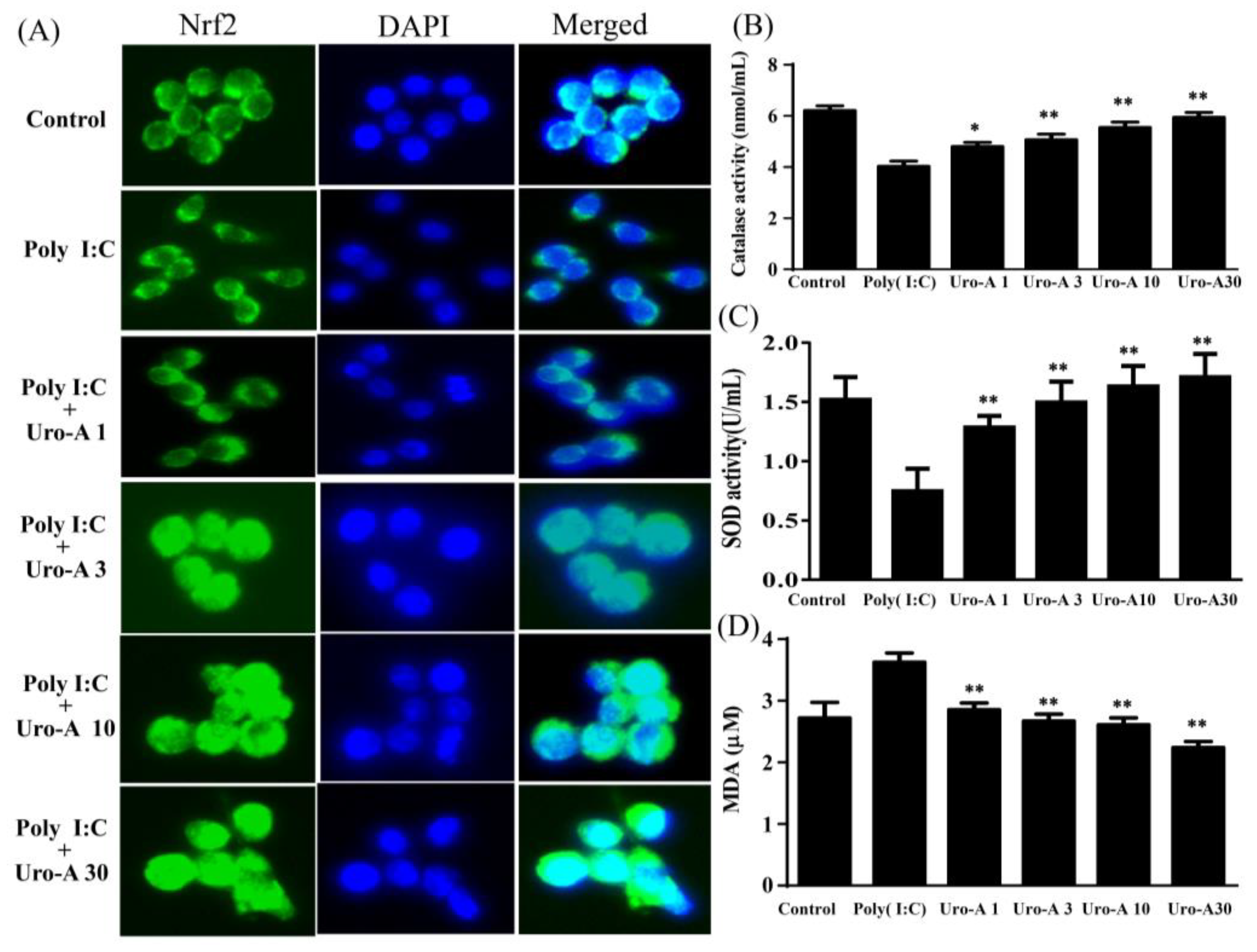
2.4. Urolithin A Elevated Nrf2 Transcriptional Regulation and Enhanced Antioxidant Cytoprotective Defense in Poly(I:C)-Stimulated RAW264.7 Cells
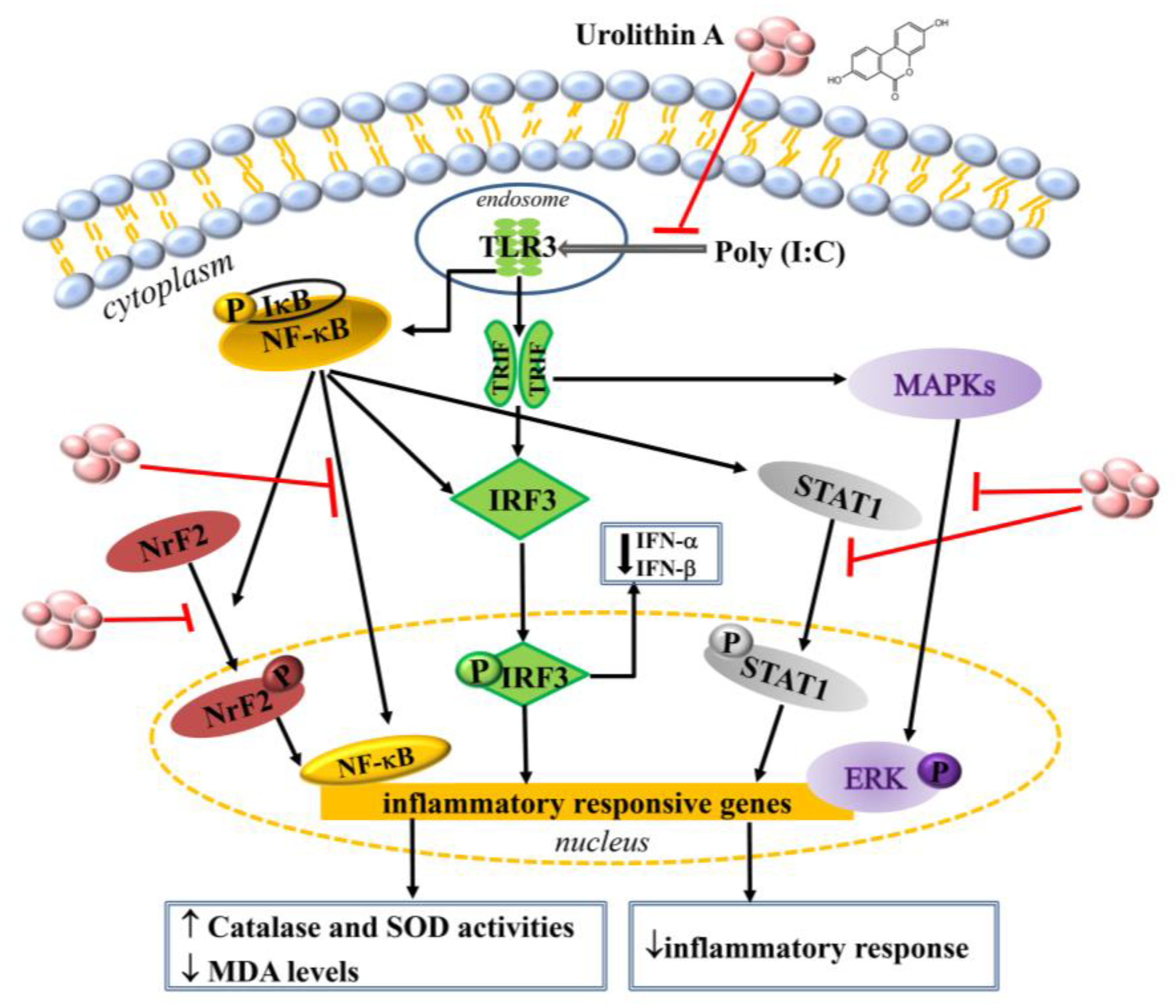
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of Urolithin A and Cell Culture
4.3. Cell Viability Assay
4.4. ELISA Assay
4.5. Preparation of Total Proteins
4.6. Western Blot Analysis
4.7. Immunofluorescence
4.8. Antioxidant Defense
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takumi, K.; Taro, K. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar]
- Takemura, N.; Kawasaki, T.; Kunisawa, J.; Sato, S.; Lamichhane, A.; Kobiyama, K.; Aoshi, T.; Ito, J.; Mizuguchi, K.; Karuppuchamy, T.; et al. Blockade of TLR3 protects mice from lethal radiation-induced gastrointestinal syndrome. Nat. Commun. 2014, 5, 3492. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Chen, C.; Niu, Z.; Gan, L.; Wang, Q.; Li, M.; Cai, X.W.; Gao, R.; Katakam, S.; Chen, H.; et al. Poly(I:C) preconditioning protects the heart against myocardial ischemia/reperfusion injury through TLR3/PI3K/Akt-dependent pathway. Signal Transduct. Target. Ther. 2020, 5, 216. [Google Scholar] [CrossRef]
- Chen, C.; Gao, R.; Li, M.; Wang, Q.; Chen, H.; Zhang, S.; Mao, X.; Behensky, A.; Zhang, Z.; Gan, L.; et al. Extracellular RNAs-TLR3 signaling contributes to cognitive decline in a mouse model of postoperative cognitive dysfunction. Brain Behav. Immun. 2019, 80, 439–451. [Google Scholar] [CrossRef]
- Janeway, C.A.; Medzhitov Jr, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Jiang, Z.; Mak, T.W.; Sen, G.; Li, X. Toll-like receptor 3-mediated activation of NF-κB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc. Natl. Acad. Sci. USA 2004, 9, 3533–3538. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, B. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Toledano, M.B.; Leonard, W.J. Modulation of transcription factor NF-κB binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 4328–4332. [Google Scholar] [CrossRef] [Green Version]
- Farzane, S.; Shikh, P.; Adity, B.; Luca, C. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 26, 5188–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chul-Su, Y.; Jwa-Jin, K.; Lee, S.J.; Hwang, J.H.; Lee, C.H.; Lee, M.S.; Jo, E.K. TLR3-Triggered Reactive Oxygen Species Contribute to Inflammatory Responses by Activating Signal Transducer and Activator of Transcription-1. J. Immunol. 2013, 15, 6368–6377. [Google Scholar]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and Mn-SOD levels in TNF alpha-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.J.; Filla, M.B.; Fultz, M.J. Autocrine/paracrine IFN-αβ mediates the lipopolysaccharide-induced activation of transcription factor Stat1α in mouse macrophages: Pivotal role of Stat1α in induction of the inducible nitric oxide synthase gene. J. Immunol. 1998, 161, 4803–4810. [Google Scholar]
- Kim, H.S.; Lee, M.S. Essential role of STAT1 in caspase-independent cell death of activated macrophages through the p38 mitogen-activated protein kinase/STAT1/reactive oxygen species pathway. Mol. Cell. Biol. 2005, 25, 6821–6833. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Shin, D.M.; Kim, K.H. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J. Immunol. 2009, 182, 3696–3705. [Google Scholar] [CrossRef]
- Moore, T.C.; Petro, T.M. IRF3 and ERK MAP-kinases control nitric oxide production from macrophages in response to poly-I:C. FEBS Lett. 2013, 587, 3014–3020. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; TenOever, B.R.; Grandvaux, N. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 30, 1148–1151. [Google Scholar] [CrossRef]
- Navarro, L.; David, M. p38-dependent activation of interferon regulatory factor 3 by lipopolysaccharide. J. Biol. Chem. 1999, 274, 35535–35538. [Google Scholar] [CrossRef] [Green Version]
- Nociari, M.; Ocheretina, O.; Murphy, M. Adenovirus induction of IRF3 occurs through a binary trigger targeting Jun N-terminal kinase and TBK1 kinase cascades and type I interferon autocrine signaling. J. Virol. 2009, 83, 4081–4091. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, M.; Chen, L.; Yang, K.; Shan, Y.; Zhu, L.; Sun, S.; Li, L.; Wang, C. The TAK1-JNK cascade is required for IRF3 function in the innate immune response. Cell Res. 2009, 19, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Anupama, M.; Rajagopal, R. Mitogen-Activated Protein Kinases and Their Role in Radiation Response. Genes Cancer 2013, 4, 401–408. [Google Scholar]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jain, A.; Gao, Y.; Dozmorov, I.M.; Mandraju, R.; Wakeland, E.K.; Pasare, C. Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc. Natl. Acad. Sci. USA 2015, 10, 13994–13999. [Google Scholar] [CrossRef] [Green Version]
- Yousif, N.M.; de Oliveira, A.C.P.; Brioschi, S.; Huell, M.; Biber, K.; Fiebich, B.L. Activation of EP2 receptor suppresses poly(I: C) and LPS-mediated inflammation in primary microglia and organotypic hippocampal slice cultures: Contributing role for MAPKs. Glia 2018, 66, 708–724. [Google Scholar] [CrossRef]
- Vučić, V. Composition and Potential Health Benefits of Pomegranate: A Review. Curr. Pharm. Des. 2019, 25, 1817–1827. [Google Scholar] [CrossRef]
- Zhao, R. Pomegranate peel polyphenols reduce chronic low-grade inflammatory responses by modulating gut microbiota and decreasing colonic tissue damage in rats fed a high-fat diet. Food Funct. 2019, 13, 8273–8285. [Google Scholar] [CrossRef]
- Begoña, C.; Paula, P.; Juan, C.E. Identification of urolithin a as a metabolite produced by human colon microflora from ellagic acid and related compounds. J. Agric. Food. Chem. 2005, 53, 5571–5576. [Google Scholar]
- Andreux, P.A.; William, B.B.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Ashley, M.T.; Darius, F.; Virginia, C.; Ramer-Tait, A.E.; Chung, S. Immunomodulatory Role of Urolithin A on Metabolic Diseases. Biomedicines 2021, 9, 192. [Google Scholar]
- Han, Q.A.; Su, D.; Shi, C.; Liu, P.; Wang, Y.; Zhuab, B.; Xia, X. Urolithin A attenuated ox-LDL-induced cholesterol accumulation in macrophages partly through regulating miR-33a and ERK/AMPK/SREBP1 signaling pathways. Food Funct. 2020, 1, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Guillermo, C.; Francisco, L.; Carmen, C.F.; Hugo, M.; López, V. The Metabolite Urolithin-A Ameliorates Oxidative Stress in Neuro-2a Cells, Becoming a Potential Neuroprotective Agent. Antioxidants 2020, 2, 177. [Google Scholar]
- Abdulrahman, A.O.; Alzubaidi, M.Y.; Nadeem, M.S.; Khan, J.A.; Rather, I.A.; Khan, M.I. The Utilization of Urolithin A—A Natural Polyphenol Metabolite of Ellagitannins as a Modulator of the Gut Microbiota for Its Potential Use in Obesity Therapy. Proceedings 2021, 79, 12. [Google Scholar]
- Tetsuya, H.; Satoshi, M.; Takashi, H.; Ohnishi, T.; Kimura, T.; Kurokawa, M.; Ieki, K.; Odaka, M.; Suzuki, S.; Watanabe, S. Cooperative Activation of CCL5 Expression by TLR3 and Tumor Necrosis Factor-α or Interferon-γ through Nuclear Factor-κB or STAT-1 in Airway Epithelial Cells. Int. Arch. Allergy Immunol. 2010, 152, 9–17. [Google Scholar]
- Laura, D.R.; Paola, G.; Francesca, P.; Soldati, L. COVID-19: Is there a role for immunonutrition in obese patient? J. Transl. Med. 2020, 18, 415. [Google Scholar]
- Dai, X.; Sayama, K.; Yamasaki, K.; Tohyama, M.; Shirakata, Y.; Hanakawa, Y.; Tokumaru, S.; Yahata, Y.; Yang, L.; Yoshimura, A. SOCS1-Negative Feedback of STAT1 Activation Is a Key Pathway in the dsRNA-Induced Innate Immune Response of Human Keratinocytes. J. Investig. Dermatol. 2006, 126, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Field, A.K.; Tytell, A.A.; Lampson, G.P. Inducers of interferon and host resistance, II. Multistranded synthetic polynucleotide complexes. Proc. Natl. Acad. Sci. USA 1967, 58, 1004–1010. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Seya, T. TLR3: Interferon induction by double-stranded RNA including poly(I:C). Adv. Drug Deliv. Rev. 2008, 7, 805–812. [Google Scholar] [CrossRef]
- Makoto, A.; Matsumoto, T.; Taguchi, K.; Kobayashi, T. Poly (I:C) impairs NO donor-induced relaxation by overexposure to NO via the NF-κB/iNOS pathway in rat superior mesenteric arteries. Free Radic. Biol. Med. 2017, 112, 553–566. [Google Scholar]
- Pinheiro de Oliveira, A.C.; Yousif, N.M.; Bhatia, H.S.; Hermanek, J.; Huell, M.; Fiebich, B.L. Poly(I:C) increases the expression of mPGES-1 and COX-2 in rat primary microglia. J. Neuroinflamm. 2016, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Maciej, C.; Zbigniew, K.; Wiktor, P.; Kochańczyk, M.; Jaruszewicz-Błońska, J.; Tudelska, K.; Błoński, S.; Kimmel, M.; Brasier, A.R.; Lipniacki, T. Cell fate in antiviral response arises in the crosstalk of IRF, NF-κB and JAK/STAT pathways. Nat. Commun. 2018, 9, 493. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, D.; Yi, L.; Lin, C.; Shi, G.; Dong, Z.; Li, J.; Fan, P.; Wang, Q.; Su, X.; Zhang, S. SARI attenuates colon inflammation by promoting STAT1 degradation in intestinal epithelial cells. Mucosal Immunol. 2019, 12, 1130–1140. [Google Scholar]
- Shawn, M.F.; Danielle, S.B.; Jason, A.S.; Athen, S.R.; Guinn, Z.P.; Pinkerton, T.S.; Petro, T.M.; Moore, T.C. MEK/ERK MAP kinase limits poly I:C-induced antiviral gene expression in RAW264.7 macrophages by reducing interferon-beta expression. FEBS Lett. 2021, 30, 2665–2674. [Google Scholar]
- Kim, H.J.; Khan, I.; Shahidullah, A.; Halimi, S.M.A.; Rauf, A.; Lee, J.Y.; Kim, Y.J.; Kim, B.Y.; Park, W. Diospyrin Modulates Inflammation in Poly I:C-Induced Macrophages via ER Stress-Induced Calcium-CHOP Pathway. Processes 2020, 8, 1050. [Google Scholar] [CrossRef]
- Nanae, H.; Takafumi, M.; Hirotsugu, U.; Harada, M. Transfection of poly(I:C) can induce reactive oxygen species-triggered apoptosis and interferon-β-mediated growth arrest in human renal cell carcinoma cells via innate adjuvant receptors and the 2-5A system. Mol. Cancer 2014, 13, 217. [Google Scholar]
- Yin, S.; Cao, W. Toll-Like Receptor Signaling Induces Nrf2 Pathway Activation through p62-Triggered Keap1 Degradation. Mol. Cell. Biol. 2015, 35, 2673–2683. [Google Scholar] [CrossRef] [Green Version]
- Shikha, M.; Damodar, G. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed. Pharm. 2018, 108, 1866–1878. [Google Scholar]
- Ahmed, N.; Nahid, S.; Naif, O.; Al-Harbi, M.M.; Ahmad, S.F. TLR-7 agonist attenuates airway reactivity and inflammation throughNrf2-mediated antioxidant protection in a murine model of allergic asthma. Int. J. Biochem. Cell. Biol. 2016, 73, 53–56. [Google Scholar]
- Marta, R.; Remzi, O.E.; Baijayanti, J.; Desponds, C.; Snäkä, T.; Prevel, F.; Isorce, N.; Lye, L.F.; Owens, K.L.; Lopes, U.G. The antioxidant response favors Leishmania parasites survival, limits inflammation and reprograms the host cell metabolism. PLoS Pathog. 2021, 25, e1009422. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.-C.; Liou, C.-J.; Shen, S.-C.; Hu, S.; Chao, J.C.-J.; Hsiao, C.-Y.; Wu, S.-J. Urolithin A Inactivation of TLR3/TRIF Signaling to Block the NF-κB/STAT1 Axis Reduces Inflammation and Enhances Antioxidant Defense in Poly(I:C)-Induced RAW264.7 Cells. Int. J. Mol. Sci. 2022, 23, 4697. https://doi.org/10.3390/ijms23094697
Huang W-C, Liou C-J, Shen S-C, Hu S, Chao JC-J, Hsiao C-Y, Wu S-J. Urolithin A Inactivation of TLR3/TRIF Signaling to Block the NF-κB/STAT1 Axis Reduces Inflammation and Enhances Antioxidant Defense in Poly(I:C)-Induced RAW264.7 Cells. International Journal of Molecular Sciences. 2022; 23(9):4697. https://doi.org/10.3390/ijms23094697
Chicago/Turabian StyleHuang, Wen-Chung, Chian-Jiun Liou, Szu-Chuan Shen, Sindy Hu, Jane C-J Chao, Chien-Yu Hsiao, and Shu-Ju Wu. 2022. "Urolithin A Inactivation of TLR3/TRIF Signaling to Block the NF-κB/STAT1 Axis Reduces Inflammation and Enhances Antioxidant Defense in Poly(I:C)-Induced RAW264.7 Cells" International Journal of Molecular Sciences 23, no. 9: 4697. https://doi.org/10.3390/ijms23094697