
Gases in Sepsis: Novel Mediators and Therapeutic Targets


Abstract
:1. Introduction
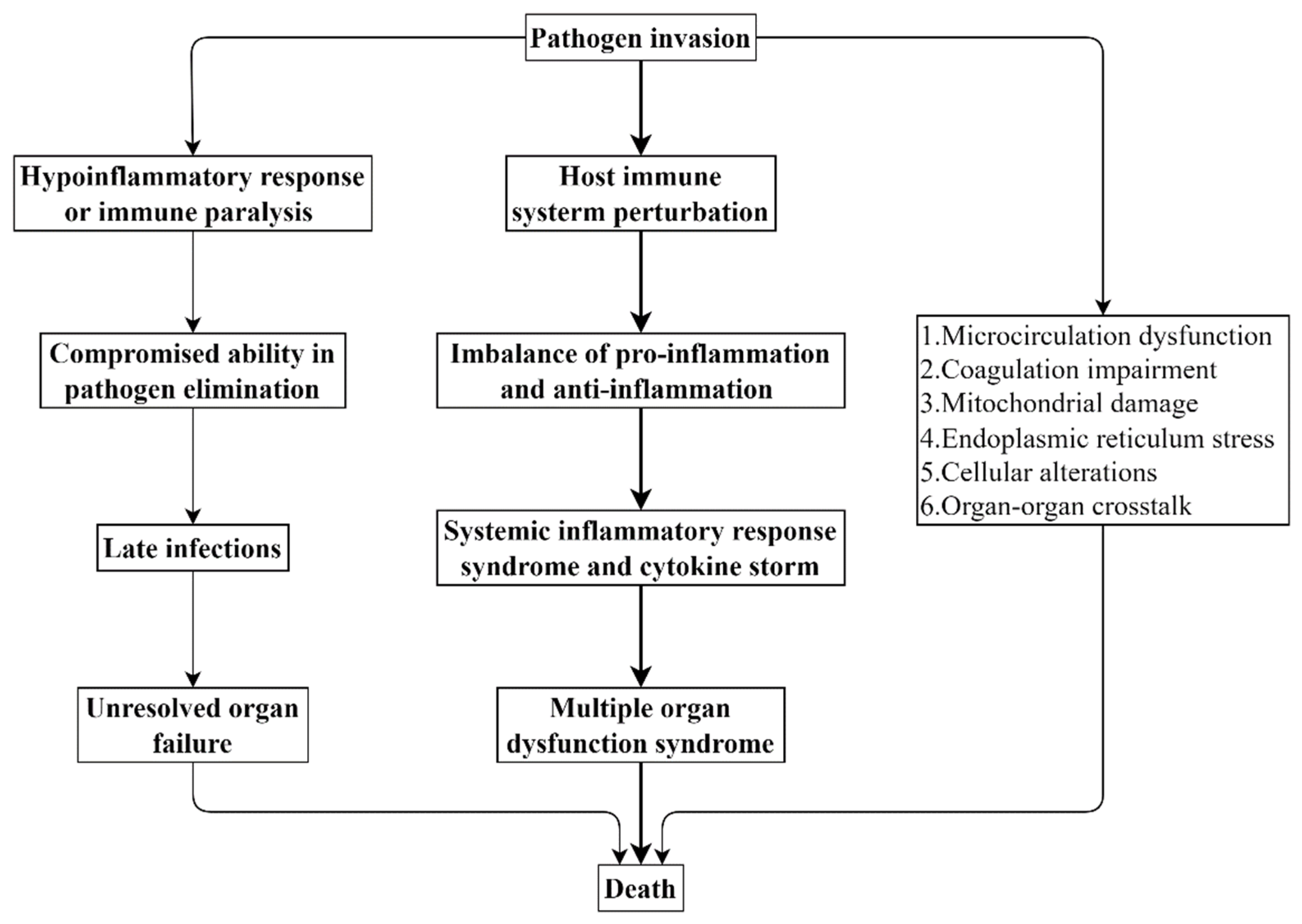
2. Pathophysiology of Sepsis
3. Gaseous Mediators
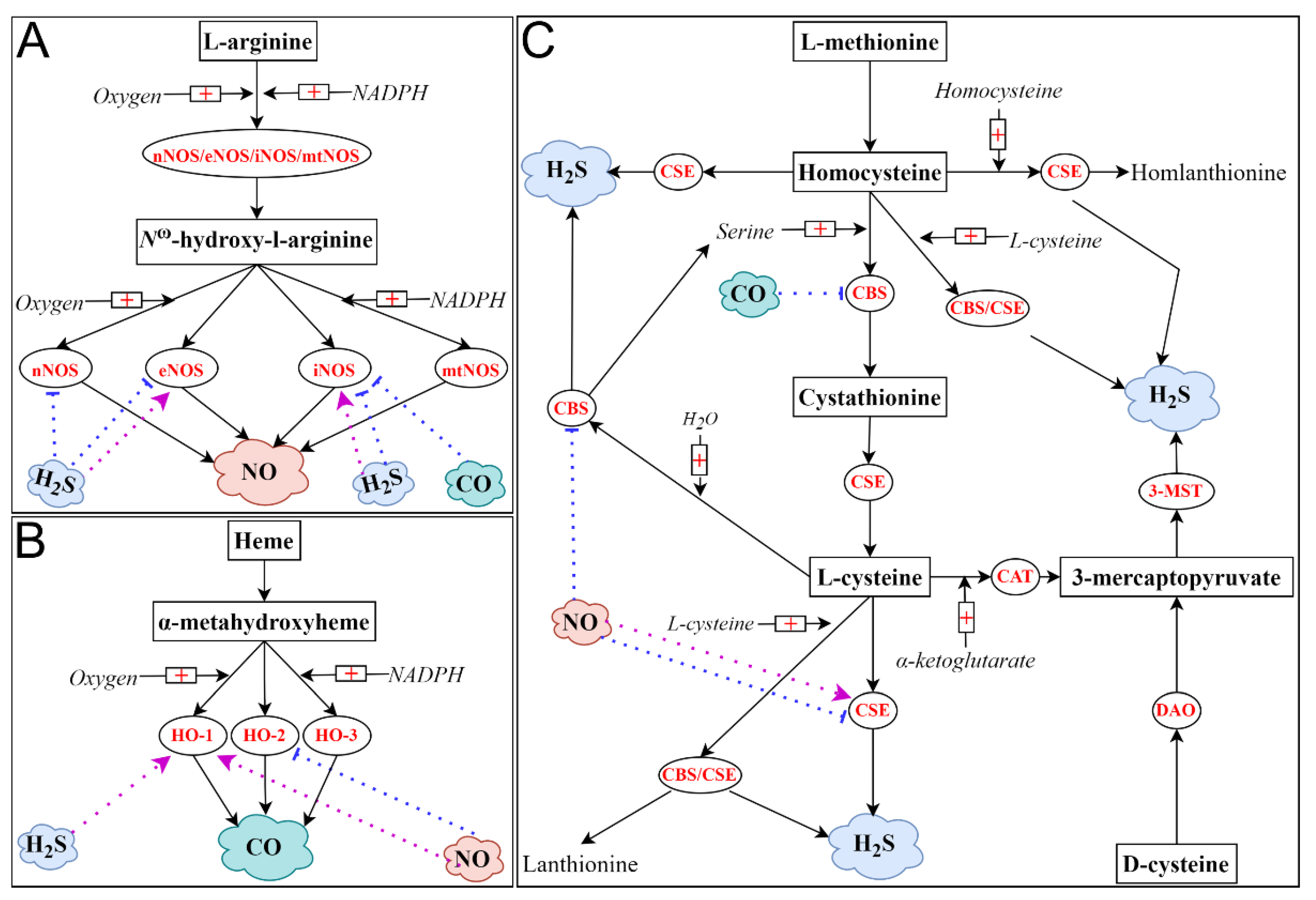
3.1. Nitric Oxide (NO)
3.2. Carbon Monoxide (CO)
3.3. Hydrogen Sulfide (H2S)
3.4. Interplay among NO, CO and H2S
4. Gaseous Mediators in Sepsis/Septic Shock
4.1. NO in Sepsis/Septic Shock
4.2. CO in Sepsis/Septic Shock
4.3. H2S in Sepsis/Septic Shock
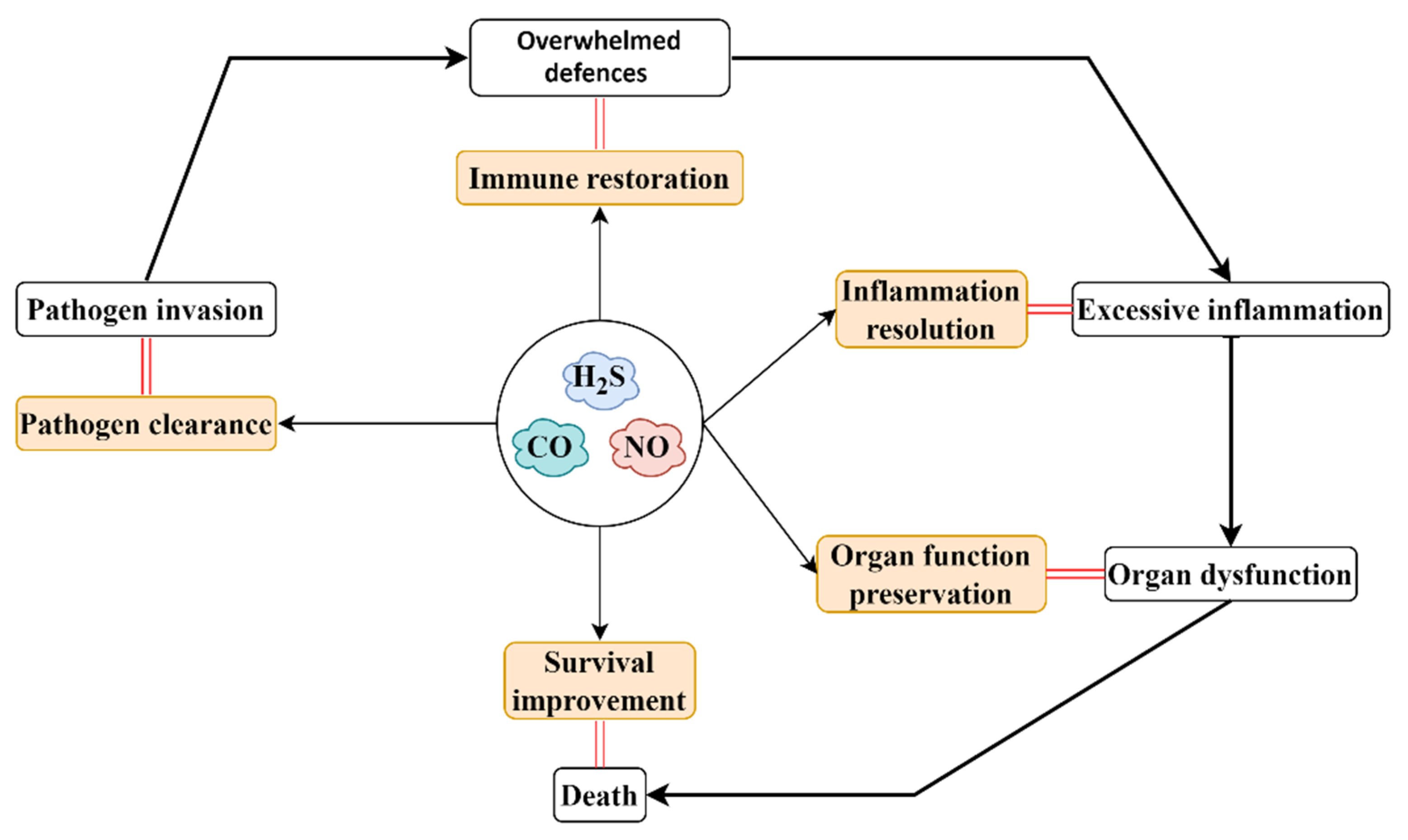
5. Gaseous-Mediator-Based Therapeutic Strategy for Sepsis/Septic Shock
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cecconi, M.; Evans, L.; Levy, M.; Rhodes, A. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Kissoon, N.; Limmathurotsakul, D.; Bory, S.; Mutahunga, B.; Seymour, C.W.; Angus, D.C.; West, T.E. The global burden of sepsis: Barriers and potential solutions. Crit. Care 2018, 22, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the global burden of disease study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, K.; Daniels, R.; Kissoon, N.; Machado, F.R.; Schachter, R.D.; Finfer, S. Recognizing sepsis as a global health priority—A WHO resolution. New Engl. J. Med. 2017, 377, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; McIntyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021, 47, 1181–1247. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Snyder, S.H. Signaling by Gasotransmitters. Sci. Signal. 2009, 2, re2. [Google Scholar] [CrossRef] [Green Version]
- Wang, R. Gasotransmitters: Growing pains and joys. Trends Biochem. Sci. 2018, 39, 283–295. [Google Scholar] [CrossRef]
- Althaus, M.; Clauss, W.G. Gasotransmitters: Novel regulators of ion channels and transporters. Front. Physiol. 2013, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.L.; Ianaro, A.; Flannigan, K.L.; Cirino, G. Gaseous mediators in resolution of inflammation. Semin. Immunol. 2015, 27, 227–233. [Google Scholar] [CrossRef]
- Hartmann, C.; Nussbaum, B.; Calzia, E.; Radermacher, P.; Wepler, M. Gaseous mediators and mitochondrial function: The future of pharmacologically induced suspended animation? Front. Physiol. 2017, 8, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2016, 15, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Kolluru, G.K.; Shen, X.; Yuan, S.; Kevil, C.G. Gasotransmitter heterocellular signaling. Antioxid. Redox Signal. 2017, 26, 936–960. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, H.; Lv, M.B.; Tang, C.; Du, J.; Jin, H. Sulfur dioxide: Endogenous generation, biological effects, detection, and therapeutic potential. Antioxid. Redox Signal. 2022, 36, 256–274. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Szabo, C. The two faces of cyanide: An environmental toxin and a potential novel mammalian gasotransmitter. FEBS J. 2021. [Google Scholar] [CrossRef]
- Boros, M.; Keppler, F. Methane production and bioactivity-a link to oxido-reductive stress. Front. Physiol. 2019, 10, 1244. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Sener, A.; Ji, Y.; Pei, Y.; Pluth, M.D. Gasotransmitters in biology and medicine: Molecular mechanisms and drug targets. Oxid. Med. Cell. Longev. 2016, 2016, 1–2. [Google Scholar] [CrossRef]
- Hendriks, K.; Maassen, H.; van Dijk, P.R.; Henning, R.; van Goor, H.; Hillebrands, J.-L. Gasotransmitters in health and disease: A mitochondria-centered view. Curr. Opin. Pharmacol. 2019, 45, 87–93. [Google Scholar] [CrossRef]
- Winkler, M.S.; Kluge, S.; Holzmann, M.; Moritz, E.; Robbe, L.; Bauer, A.; Zahrte, C.; Priefler, M.; Schwedhelm, E.; Böger, R.H.; et al. Markers of nitric oxide are associated with sepsis severity: An observational study. Crit. Care 2017, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Choi, A.M.K. Carbon monoxide in the treatment of sepsis. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1387–L1393. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Teng, X.; Hu, Z.-J.; Tian, D.-Y.; Jin, S.; Wu, Y.-M. Hydrogen sulfide attenuated sepsis-induced myocardial dysfunction through TLR4 pathway and endoplasmic reticulum stress. Front. Physiol. 2021, 12, 653601. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, C.; Jaimes, F. Organ dysfunction in sepsis: An ominous trajectory from infection to death. Yale J. Biol Med. 2019, 92, 629–640. [Google Scholar] [PubMed]
- Pool, R.; Gomez, H.; Kellum, J.A. Mechanisms of organ dysfunction in sepsis. Crit. Care Clin. 2018, 34, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Lelubre, C.; Vincent, J.-L. Mechanisms and treatment of organ failure in sepsis. Nat. Rev. Nephrol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Huang, M.; Cai, S.; Su, J. The pathogenesis of sepsis and potential therapeutic targets. Int. J. Mol. Sci. 2019, 20, 5376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delano, M.J.; Ward, P.A. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol. Rev. 2016, 274, 330–353. [Google Scholar] [CrossRef] [PubMed]
- Conway-Morris, A.; Wilson, J.; Shankar-Hari, M. Immune activation in sepsis. Crit. Care Clin. 2018, 34, 29–42. [Google Scholar] [CrossRef]
- Lawal, B.; Wang, Y.-C.; Wu, A.T.H.; Huang, H.-S. Pro-Oncogenic c-Met/EGFR, biomarker signatures of the tumor microenvironment are clinical and therapy response prognosticators in colorectal cancer, and therapeutic targets of 3-Phenyl-2H-benzo[e][1,3]-Oxazine-2,4(3H)-Dione derivatives. Front. Pharmacol. 2021, 12, 691234. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Jarczak, D.; Kluge, S.; Nierhaus, A. Sepsis—Pathophysiology and therapeutic concepts. Front. Med. 2021, 8, 628302. [Google Scholar] [CrossRef] [PubMed]
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Cao, S.; Zhou, Y.; Xiong, Y. Recent advances in endotoxin tolerance. J. Cell Biochem. 2019, 120, 56–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamers, L.; Kox, M.; Pickkers, P. Sepsis-induced immunoparalysis: Mechanisms, markers, and treatment options. Minerva Anestesiol 2014, 81, 426–439. [Google Scholar] [PubMed]
- Venet, F.; Monneret, G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol. 2018, 14, 121–137. [Google Scholar] [CrossRef]
- Chen, J.; Wei, H. Immune intervention in sepsis. Front. Pharmacol. 2021, 12, 718089. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric oxide in cellular adaptation and disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef] [PubMed]
- Król, M.; Kepinska, M. Human nitric oxide Synthase—Its functions, polymorphisms, and inhibitors in the context of inflammation, diabetes and cardiovascular diseases. Int. J. Mol. Sci. 2020, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Dynnik, V.V.; Grishina, E.V.; Fedotcheva, N.I. The mitochondrial NO-synthase/guanylate cyclase/protein kinase G signaling system underpins the dual effects of nitric oxide on mitochondrial respiration and opening of the permeability transition pore. FEBS J. 2019, 287, 1525–1536. [Google Scholar] [CrossRef]
- DeMartino, A.W.; Kim-Shapiro, D.B.; Patel, R.P.; Gladwin, M.T. Nitrite and nitrate chemical biology and signalling. Br. J. Pharmacol. 2019, 176, 228–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapil, V.; Khambata, R.S.; Jones, D.A.; Rathod, K.; Primus, C.; Massimo, G.; Fukuto, J.M.; Ahluwalia, A. The noncanonical pathway for in vivo nitric oxide generation: The nitrate-nitrite-nitric oxide pathway. Pharmacol. Rev. 2020, 72, 692–766. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, A.; Kowalska, M.; Nowaczyk, J.; Grześk, G. Carbon monoxide and nitric oxide as examples of the youngest class of transmitters. Int. J. Mol. Sci. 2021, 22, 6029. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J. The identification of nitric oxide as endothelium-derived relaxing factor. Circ. Res. 2013, 113, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Cirino, G.; Vellecco, V.; Bucci, M. Nitric oxide and hydrogen sulfide: The gasotransmitter paradigm of the vascular system. J. Cereb. Blood Flow Metab. 2017, 174, 4021–4031. [Google Scholar] [CrossRef] [Green Version]
- Farah, C.; Michel, L.Y.; Balligand, J.-L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef]
- Ghimire, K.; Altmann, H.M.; Straub, A.C.; Isenberg, J.S. Nitric oxide: What’s new to NO? Am. J. Physiol. Cell Physiol. 2017, 312, C254–C262. [Google Scholar] [CrossRef]
- Shah, S.; Karathanasi, A.; Revythis, A.; Ioannidou, E.; Boussios, S. Cancer-Associated thrombosis: A new light on an old story. Diseases 2021, 9, 34. [Google Scholar] [CrossRef]
- Fagone, P.; Mazzon, E.; Bramanti, P.; Bendtzen, K.; Nicoletti, F. Gasotransmitters and the immune system: Mode of action and novel therapeutic targets. Eur. J. Pharmacol. 2018, 834, 92–102. [Google Scholar] [CrossRef]
- Kuschman, H.P.; Palczewski, M.B.; Thomas, D.D. Nitric oxide and hydrogen sulfide: Sibling rivalry in the family of epigenetic regulators. Free Radic. Biol. Med. 2021, 170, 34–43. [Google Scholar] [CrossRef]
- McGinity, C.; Palmieri, E.; Somasundaram, V.; Bhattacharyya, D.; Ridnour, L.; Cheng, R.; Ryan, A.; Glynn, S.; Thomas, D.; Miranda, K.; et al. Nitric oxide modulates metabolic processes in the tumor immune microenvironment. Int. J. Mol. Sci. 2021, 22, 7068. [Google Scholar] [CrossRef] [PubMed]
- Opatrilova, R.; Kubatka, P.; Caprnda, M.; Büsselberg, D.; Krasnik, V.; Veselý, P.; Saxena, S.; Ruia, S.; Mozos, I.; Rodrigo, L.; et al. Nitric oxide in the pathophysiology of retinopathy: Evidences from preclinical and clinical researches. Acta Ophthalmol. 2018, 96, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Hopper, C.P.; Zambrana, P.N.; Goebel, U.; Wollborn, J. A brief history of carbon monoxide and its therapeutic origins. Nitric. Oxide 2021, 111-112, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, R. Carbon monoxide: Endogenous production, physiological functions, and pharmacological applications. Pharmacol. Rev. 2005, 57, 585–630. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of heme oxygenase as a modulator of heme-mediated pathways. Antioxidants 2019, 8, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olas, B. Carbon monoxide is not always a poison gas for human organism: Physiological and pharmacological features of CO. Chem. Interact. 2014, 222, 37–43. [Google Scholar] [CrossRef]
- Sethi, J.M. Carbon monoxide. Crit. Care Med. 2005, 33, S496–S497. [Google Scholar] [CrossRef]
- Gullotta, F.; di Masi, A.; Coletta, M.; Ascenzi, P. CO metabolism, sensing, and signaling. BioFactors 2011, 38, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Hsu, A.; Moore, P.K. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation—a tale of three gases! Pharmacol. Ther. 2009, 123, 386–400. [Google Scholar] [CrossRef]
- Ameredes, B.T.; Otterbein, L.E.; Kohut, L.K.; Gligonic, A.L.; Calhoun, W.J.; Choi, A.M.K. Low-dose carbon monoxide reduces airway hyperresponsiveness in mice. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L1270–L1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Głowacka, U.; Brzozowski, T.; Magierowski, M. Synergisms, discrepancies and interactions between hydrogen sulfide and carbon monoxide in the gastrointestinal and digestive system physiology, pathophysiology and pharmacology. Biomolecules 2020, 10, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef] [Green Version]
- Takaki, S.; Takeyama, N.; Kajita, Y.; Yabuki, T.; Noguchi, H.; Miki, Y.; Inoue, Y.; Nakagawa, T.; Noguchi, H. Beneficial effects of the heme oxygenase-1/carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med. 2010, 36, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Ma, K.C.; Choi, A.M.K. Carbon monoxide in lung cell physiology and disease. Am. J. Physiol. Physiol. 2018, 314, C211–C227. [Google Scholar] [CrossRef] [PubMed]
- Owens, E.O. Endogenous carbon monoxide production in disease. Clin. Biochem. 2010, 43, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular regulation and pharmacological inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Szabo, C. A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharmacol. 2018, 149, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. H2S and polysulfide metabolism: Conventional and unconventional pathways. Biochem. Pharmacol. 2018, 149, 77–90. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxidants Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H. Hydrogen sulfide: Its production and functions. Exp. Physiol. 2011, 96, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. The physiological role of hydrogen sulfide and beyond. Nitric Oxide 2014, 41, 4–10. [Google Scholar] [CrossRef]
- Rose, P.; Moore, P.K.; Zhu, Y.Z. H2S biosynthesis and catabolism: New insights from molecular studies. Cell. Mol. Life Sci. 2017, 74, 1391–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, M. Hydrogen sulfide and substance P in inflammation. Antioxid. Redox Signal. 2010, 12, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide (H(2)S) and polysulfide (H(2)S(n)) signaling: The first 25 years. Biomolecules 2021, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [Green Version]
- Wang, R. Hydrogen sulfide: A new EDRF. Kidney Int. 2009, 76, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Ping, N.-N.; Li, S.; Mi, Y.-N.; Cao, L.; Cao, Y.-X. Hydrogen sulphide induces vasoconstriction of rat coronary artery via activation of Ca 2+ influx. Acta Physiol. 2015, 214, 88–96. [Google Scholar] [CrossRef]
- Ali, M.Y.; Ping, C.Y.; Mok, Y.Y.; Ling, L.; Whiteman, M.; Bhatia, M.; Moore, P.K. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 2006, 149, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Lv, B.; Chen, S.; Tang, C.; Jin, H.; Du, J.; Huang, Y. Hydrogen sulfide and vascular regulation–An update. J. Adv. Res. 2021, 27, 85–97. [Google Scholar] [CrossRef]
- Khattak, S.; Zhang, Q.-Q.; Sarfraz, M.; Muhammad, P.; Ngowi, E.; Khan, N.; Rauf, S.; Wang, Y.-Z.; Qi, H.-W.; Wang, D.; et al. The role of hydrogen sulfide in respiratory diseases. Biomolecules 2021, 11, 682. [Google Scholar] [CrossRef]
- Madurga, A.; Golec, A.; Pozarska, A.; Ishii, I.; Mižíková, I.; Nardiello, C.; Vadász, I.; Herold, S.; Mayer, K.; Reichenberger, F.; et al. The H2S-generating enzymes cystathionine β-synthase and cystathionine γ-lyase play a role in vascular development during normal lung alveolarization. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L710–L724. [Google Scholar] [CrossRef] [PubMed]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Shefa, U.; Kim, M.-S.; Jeong, N.Y.; Jung, J. Antioxidant and cell-signaling functions of hydrogen sulfide in the central nervous system. Oxidative Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Manandhar, S.; Sinha, P.; Ejiwale, G.; Bhatia, M. Hydrogen sulfide and its interaction with other players in inflammation. Adv. Exp. Med. Biol. 2021, 1315, 129–159. [Google Scholar] [CrossRef]
- Sun, F.; Luo, J.; Yue, T.; Wang, F.; Yang, C.; Zhang, J.; Wang, X.; Wang, C. The role of hydrogen sulphide signalling in macrophage activation. Immunology 2021, 162, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Jin, H.; Yang, L. Role of hydrogen sulfide in retinal diseases. Front. Pharmacol. 2017, 8, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Teng, X.; Jin, S.; Wu, Y. The antiviral roles of hydrogen sulfide by blocking the interaction between SARS-CoV-2 and its potential cell surface receptors. Oxid. Med. Cell Longev. 2021, 2021, 7866992. [Google Scholar] [CrossRef] [PubMed]
- Pae, H.-O.; Lee, Y.C.; Jo, E.-K.; Chung, H.-T. Subtle interplay of endogenous bioactive gases (NO, CO and H2S) in inflammation. Arch. Pharmacal Res. 2009, 32, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Nilius, B.; Han, J. Gaseous signaling molecules in cardiovascular function: From mechanisms to clinical translation. Rev. Physiol. Biochem. Pharmacol. 2018, 174, 81–156. [Google Scholar] [CrossRef]
- Liew, H.; Khoo, H.; Moore, P.; Bhatia, M.; Lu, J.; Moochhala, S. Synergism between hydrogen sulfide (H2S) and nitric oxide (NO) in vasorelaxation induced by stonustoxin (SNTX), a lethal and hypotensive protein factor isolated from stonefish Synanceja horrida venom. Life Sci. 2007, 80, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Kostetski, I.; Chu, S.H.; Siau, J.L.; Bhatia, M.; Moore, P.K. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun. 2006, 343, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.T.; Choi, B.M.; Kwon, Y.G.; Kim, Y.M. Interactive relations between nitric oxide (NO) and carbon monoxide (CO): Heme oxygenase-1/CO pathway is a key modulator in NO-mediated antiapoptosis and anti-inflammation. Methods Enzym. 2008, 441, 329–338. [Google Scholar]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on Hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxidative Med. Cell. Longev. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Vicente, J.B.; Colaço, H.G.; Mendes, M.I.; Sarti, P.; Leandro, P.; Giuffrè, A. NO* binds human cystathionine β-synthase quickly and tightly. J. Biol. Chem. 2014, 289, 8579–8587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. Embo. J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, S.; Fledderus, J.O.; Verhaar, M.; Joles, J.A. Beneficial effects of diminished production of hydrogen sulfide or carbon monoxide on hypertension and renal injury induced by NO withdrawal. Br. J. Pharmacol. 2015, 172, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-H.; Xue, W.-L.; Wang, M.-J.; Zhou, Y.; Zhang, C.-C.; Sun, C.; Zhu, L.; Liang, K.; Chen, Y.; Tao, B.-B.; et al. H2S regulates endothelial nitric oxide synthase protein stability by promoting microRNA-455-3p expression. Sci. Rep. 2017, 7, 44807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heine, C.L.; Schmidt, R.; Geckl, K.; Schrammel, A.; Gesslbauer, B.; Schmidt, K.; Mayer, B.; Gorren, A.C.F. Selective irreversible inhibition of neuronal and inducible nitric-oxide synthase in the combined presence of hydrogen sulfide and nitric oxide. J. Biol. Chem. 2015, 290, 24932–24944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, W.; Chen, Q.; Gong, F.; Xie, C.; Zhou, S.; Gao, L. Cardioprotection of H2S by downregulating iNOS and upregulating HO-1 expression in mice with CVB3-induced myocarditis. Life Sci. 2013, 93, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.O.; Pae, H.O.; Oh, G.S.; Jeong, G.S.; Lee, B.S.; Lee, S.; Kim, D.Y.; Rhew, H.Y.; Lee, K.M.; Chung, H.T. Hydrogen sulfide potentiates interleukin-1beta-induced nitric oxide production via enhancement of extracellular signal-regulated kinase activation in rat vascular smooth muscle cells. Biochem. Biophys Res. Commun. 2006, 345, 938–944. [Google Scholar] [CrossRef] [PubMed]
- De Cruz, S.J.; Kenyon, N.J.; Sandrock, C.E. Bench-to-bedside review: The role of nitric oxide in sepsis. Expert Rev. Respir. Med. 2009, 3, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Cinel, I. Bench-to-bedside review: Nitric oxide in critical illness–update 2008. Crit. Care 2009, 13, 218–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-C.; Ebihara, S.; EL Dwairi, Q.; Hussain, S.N.A.; Yang, L.; Gottfried, S.B.; Comtois, A.; Petrof, B.J. Diaphragm Sarcolemmal injury is induced by sepsis and alleviated by nitric oxide synthase inhibition. Am. J. Respir. Crit. Care Med. 1998, 158, 1656–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkharfy, K.M.; Ahmad, A.; Raish, M.; Vanhoutte, P.M. Thymoquinone modulates nitric oxide production and improves organ dysfunction of sepsis. Life Sci. 2015, 143, 131–138. [Google Scholar] [CrossRef]
- Luo, M.; Luo, S.; Cheng, Z.; Yang, X.; Lv, D.; Li, X.; Guo, Y.; Li, C.; Yan, J. Tubeimoside I improves survival of mice in sepsis by inhibiting inducible nitric oxide synthase expression. Biomed. Pharmacother. 2020, 126, 110083. [Google Scholar] [CrossRef]
- Yu, M.-H.; Chen, M.-H.; Han, F.; Li, Q.; Sun, R.-H.; Tu, Y.-X. Prognostic value of the biomarkers serum amyloid A and nitric oxide in patients with sepsis. Int. Immunopharmacol. 2018, 62, 287–292. [Google Scholar] [CrossRef]
- Lambden, S. Bench to bedside review: Therapeutic modulation of nitric oxide in sepsis—an update. Intensiv. Care Med. Exp. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauwels, A. Nitric oxide in shock. Kidney Int. 2007, 72, 557–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Besch, V.; Khaibullina, A.; Hergen, A.; Quezado, M.; Eichacker, P.; Quezado, Z.M.N. Neuronal nitric oxide synthase deficiency decreases survival in bacterial peritonitis and sepsis. Intensiv. Care Med. 2007, 33, 1993–2003. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Pathak, S.; Sarikhani, M.; Majumdar, S.; Ray, S.; Chandrasekar, B.S.; Adiga, V.; Sundaresan, N.R.; Nandi, D. Nitric oxide synthase 2 enhances the survival of mice during Salmonella Typhimurium infection-induced sepsis by increasing reactive oxygen species, inflammatory cytokines and recruitment of neutrophils to the peritoneal cavity. Free Radic. Biol. Med. 2018, 116, 73–87. [Google Scholar] [CrossRef]
- Hoetzel, A.; Dolinay, T.; Schmidt, R.; Choi, A.M.K.; Ryter, S.W. Carbon monoxide in sepsis. Antioxid. Redox Signal. 2007, 9, 2013–2026. [Google Scholar] [CrossRef] [PubMed]
- Kyokane, T.; Norimizu, S.; Taniai, H.; Yamaguchi, T.; Takeoka, S.; Tsuchida, E.; Naito, M.; Nimura, Y.; Ishimura, Y.; Suematsu, M. Carbon monoxide from heme catabolism protects against hepatobiliary dysfunction in endotoxin-treated rat liver. Gastroenterology 2001, 120, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, M.A. Heme oxygenase-1–derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Investig. 2008, 118, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoyi, K.; Lee, T.Y.; Lee, Y.S.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. Heme-Oxygenase-1 Induction and Carbon Monoxide-Releasing Molecule Inhibit Lipopolysaccharide (LPS)-Induced High-Mobility Group Box 1 Release in Vitro and Improve Survival of Mice in LPS- and Cecal Ligation and Puncture-Induced Sepsis Model in Vivo. Mol. Pharmacol. 2009, 76, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adach, W.; Błaszczyk, M.; Olas, B. Carbon monoxide and its donors-Chemical and biological properties. Chem. Interact. 2020, 318, 108973. [Google Scholar] [CrossRef]
- Zhang, W.; Tao, A.; Lan, T.; Cepinskas, G.; Kao, R.; Martin, C.M.; Rui, T. Carbon monoxide releasing molecule-3 improves myocardial function in mice with sepsis by inhibiting NLRP3 inflammasome activation in cardiac fibroblasts. Basic Res. Cardiol. 2017, 112, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, S.; Zhao, H.; Qin, H.; Zhang, J.; Dong, J.; Zhang, H.; Liu, X.; Zhao, Z.; Zhao, Y.; et al. Carbon monoxide inhibits the expression of proteins associated with intestinal mucosal Pyroptosis in a rat model of sepsis induced by Cecal ligation and puncture. Med. Sci. Monit. 2020, 26, e920668-1. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Larsen, R.; Gallo, D.; Chin, B.Y.; Harris, C.; Mannam, P.; Kaczmarek, E.; Lee, P.J.; Zuckerbraun, B.S.; Flavell, R.; et al. Macrophages sense and kill bacteria through carbon monoxide–dependent inflammasome activation. J. Clin. Investig. 2014, 124, 4926–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Huang, J.; Li, Y.; Chang, R.; Wu, H.; Lin, J.; Huang, Z. Exogenous carbon monoxide decreases sepsis-induced acute kidney injury and inhibits NLRP3 inflammasome activation in rats. Int. J. Mol. Sci. 2015, 16, 20595–20608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Motterlini, R.; Neviere, R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2009, 329, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacGarvey, N.C.; Suliman, H.B.; Bartz, R.R.; Fu, P.; Withers, C.M.; Welty-Wolf, K.E.; Piantadosi, C.A. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 2012, 185, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unuma, K.; Aki, T.; Nagano, S.; Watanabe, R.; Uemura, K. The down-regulation of cardiac contractile proteins underlies myocardial depression during sepsis and is mitigated by carbon monoxide. Biochem. Biophys. Res. Commun. 2018, 495, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Zhang, J.; Lv, W.; Wang, X.; Sun, B. Effect of carbon monoxide-releasing molecules II-liberated CO on suppressing inflammatory response in sepsis by interfering with nuclear factor kappa B activation. PLoS ONE 2013, 8, e75840. [Google Scholar] [CrossRef]
- Riquelme, S.A.; Bueno, S.M.; Kalergis, A.M. Carbon monoxide down-modulates Toll-like receptor 4/MD2 expression on innate immune cells and reduces endotoxic shock susceptibility. Immunology 2015, 144, 321–332. [Google Scholar] [CrossRef]
- Liu, D.; Wang, X.; Qin, W.; Chen, J.; Wang, Y.; Zhuang, M.; Sun, B. Suppressive effect of exogenous carbon monoxide on endotoxin-stimulated platelet over-activation via the glycoprotein-mediated PI3K-Akt-GSK3β pathway. Sci. Rep. 2016, 6, 23653. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Du, R.; Yin, S.; Liu, X.; Xu, G.; Cao, W. Nrf2 is essential for the anti-inflammatory effect of carbon monoxide in LPS-induced inflammation. Inflamm. Res. 2015, 64, 537–548. [Google Scholar] [CrossRef]
- Tsoyi, K.; Hall, S.R.R.; Dalli, J.; Colas, R.A.; Ghanta, S.; Ith, B.; Coronata, A.; Fredenburgh, L.E.; Baron, R.M.; Choi, A.M.K.; et al. Carbon monoxide improves efficacy of mesenchymal stromal cells during sepsis by production of specialized proresolving lipid mediators*. Crit. Care Med. 2016, 44, e1236–e1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegdi, R.; Perrin, D.; Burdin, M.; Boiteau, R.; Tenaillon, A. Increased endogenous carbon monoxide production in severe sepsis. Intensiv. Care Med. 2002, 28, 793–796. [Google Scholar] [CrossRef]
- Iwasashi, H.; Suzuki, M.; Unno, M.; Utiyama, T.; Oikawa, M.; Kondo, N.; Matsuno, S. Inhibition of heme oxygenase ameliorates sepsis-induced liver dysfunction in rats. Surg. Today 2003, 33, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Du, J.; Tang, C.; Bin, G.; Jiang, H. Changes in arterial hydrogen sulfide (H2S) content during septic shock and endotoxin shock in rats. J. Infect. 2003, 47, 155–160. [Google Scholar] [CrossRef]
- Zhang, H.; Zhi, L.; Moore, P.K.; Bhatia, M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L1193–L1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhi, L.; Moochhala, S.; Moore, P.K.; Bhatia, M. Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-kappaB. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L960–L971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhi, L.; Moochhala, S.M.; Moore, P.K.; Bhatia, M. Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J. Leukoc. Biol. 2007, 82, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Moochhala, S.M.; Bhatia, M. Endogenous hydrogen sulfide regulates inflammatory response by activating the ERK pathway in polymicrobial sepsis. J. Immunol. 2008, 181, 4320–4331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hegde, A.; Ng, S.W.; Adhikari, S.; Moochhala, S.M.; Bhatia, M. Hydrogen sulfide up-regulates substance P in polymicrobial sepsis-associated lung injury. J. Immunol. 2007, 179, 4153–4160. [Google Scholar] [CrossRef] [Green Version]
- Ang, S.-F.; Moochhala, S.M.; Bhatia, M. Hydrogen sulfide promotes transient receptor potential vanilloid 1-mediated neurogenic inflammation in polymicrobial sepsis*. Crit. Care Med. 2010, 38, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.F.; Moochhala, S.M.; MacAry, P.A.; Bhatia, M. Hydrogen sulfide and neurogenic inflammation in polymicrobial sepsis: Involvement of substance P and ERK-NF-κB signaling. PLoS ONE 2011, 6, e24535. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.-F.; Sio, S.W.S.; Moochhala, S.M.; Macary, P.A.; Bhatia, M. Hydrogen Sulfide Upregulates Cyclooxygenase-2 and Prostaglandin E Metabolite in Sepsis-Evoked Acute Lung Injury via Transient Receptor Potential Vanilloid Type 1 Channel Activation. J. Immunol. 2011, 187, 4778–4787. [Google Scholar] [CrossRef] [PubMed]
- Badiei, A.; Chambers, S.; Gaddam, R.R.; Bhatia, M. Cystathionine-γ-lyase gene silencing with siRNA in monocytes/macrophages attenuates inflammation in cecal ligation and puncture-induced sepsis in the mouse. J. Biosci. 2016, 41, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Norris, E.J.; Feilen, N.; Nguyen, N.H.; Culberson, C.R.; Shin, M.C.; Fish, M.; Clemens, M.G. Hydrogen sulfide modulates sinusoidal constriction and contributes to hepatic microcirculatory dysfunction during endotoxemia. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1070–G1078. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.M.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Gaddam, R.R.; Fraser, R.; Badiei, A.; Chambers, S.; Cogger, V.C.; Le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase Gene Deletion Protects Mice against Inflammation and Liver Sieve Injury following Polymicrobial Sepsis. PLoS ONE 2016, 11, e0160521. [Google Scholar] [CrossRef]
- Gaddam, R.R.; Chambers, S.; Fraser, R.; Cogger, V.C.; Le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase-Derived hydrogen sulfide-regulated substance P modulates liver sieve fenestrations in Caecal ligation and puncture-induced sepsis. Int. J. Mol. Sci. 2019, 20, 3191. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, X.; Pan, L.; Wu, W.; Yang, D.; Qin, M.; Jia, W.; Xiao, C.; Long, F.; Ge, J.; et al. Endogenous hydrogen sulfide regulates histone demethylase JMJD3-mediated inflammatory response in LPS-stimulated macrophages and in a mouse model of LPS-induced septic shock. Biochem. Pharmacol. 2018, 149, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Gaddam, R.R.; Chambers, S.; Murdoch, D.; Shaw, G.; Bhatia, M. Circulating levels of hydrogen sulfide and substance P in patients with sepsis. J. Infect. 2017, 75, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Bee, N.; White, R.; Petros, A.J. Hydrogen sulfide in exhaled gases from ventilated septic neonates and children: A preliminary report. Pediatr. Crit. Care Med. 2017, 18, e327–e332. [Google Scholar] [CrossRef]
- Spiller, F.; Orrico, M.I.; Nascimento, D.C.; Czaikoski, P.G.; Souto, F.O.; Alves-Filho, J.C.; Freitas, A.; Carlos, D.; Montenegro, M.F.; Neto, A.F.; et al. Hydrogen sulfide improves neutrophil migration and survival in sepsis via K+ATP channel activation. Am. J. Respir. Crit. Care Med. 2010, 182, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Druzhyna, N.; Szabo, C. Delayed Treatment with sodium hydrosulfide improves regional blood flow and alleviates Cecal ligation and puncture (CLP)-Induced septic shock. Shock 2016, 46, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-X.; Du, J.-M.; Ding, Z.-N.; Zhu, X.-Y.; Jiang, L.; Liu, Y.-J. Hydrogen sulfide prevents diaphragm weakness in Cecal ligation puncture-induced sepsis by preservation of mitochondrial function. Am. J. Transl. Res. 2017, 9, 3270–3281. [Google Scholar] [PubMed]
- Li, J.; Ma, J.; Li, M.; Tao, J.; Chen, J.; Yao, C.; Yao, S. GYY4137 alleviates sepsis-induced acute lung injury in mice by inhibiting the PDGFRβ/Akt/NF-κB/NLRP3 pathway. Life Sci. 2021, 271, 119192. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Li, L.; Ma, J.; Yao, C.; Yao, S. Hydrogen sulfide attenuates ferroptosis and stimulates autophagy by blocking mTOR signaling in sepsis-induced acute lung injury. Mol. Immunol. 2021, 141, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Chen, X.; Luo, Z.; Zhao, L.; Zhang, T.; Yang, N.; Long, X.; Xie, H.; Liu, J.; Xu, W. Inhibition of endogenous hydrogen sulfide production exacerbates the inflammatory response during urine-derived sepsis-induced kidney injury. Exp. Ther. Med. 2018, 16, 2851–2858. [Google Scholar] [CrossRef]
- Renieris, G.; Droggiti, D.-E.; Katrini, K.; Koufargyris, P.; Gkavogianni, T.; Karakike, E.; Antonakos, N.; Damoraki, G.; Karageorgos, A.; Sabracos, L.; et al. Host cystathionine-γ lyase derived hydrogen sulfide protects against Pseudomonas aeruginosa sepsis. PLOS Pathog. 2021, 17, e1009473. [Google Scholar] [CrossRef] [PubMed]
- Aslami, H.; Pulskens, W.P.; Kuipers, M.T.; Bos, A.P.; Van Kuilenburg, A.B.P.; Wanders, R.J.A.; Roelofsen, J.; Roelofs, J.; Kerindongo, R.P.; Beurskens, C.J.P.; et al. Hydrogen sulfide donor NaHS reduces organ injury in a rat model of pneumococcal pneumosepsis, associated with improved bio-energetic status. PLoS ONE 2013, 8, e63497. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Chen, J.; Yu, F.; Liu, W.; He, M. GYY4137 protected the integrity of the blood-brain barrier via activation of the Nrf2/ARE pathway in mice with sepsis. FASEB J. 2021, 35, e21710. [Google Scholar] [CrossRef] [PubMed]
- Harbrecht, B.G. Therapeutic use of nitric oxide scavengers in shock and sepsis. Curr. Pharm. Des. 2006, 12, 3543–3549. [Google Scholar] [CrossRef]
- Teman, N.R.; Thomas, J.; Bryner, B.S.; Haas, C.F.; Haft, J.W.; Park, P.K.; Lowell, M.J.; Napolitano, L.M. Inhaled nitric oxide to improve oxygenation for safe critical care transport of adults with severe hypoxemia. Am. J. Crit. Care 2015, 24, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, S.; Glaspey, L.J.; Dellinger, R.P.; Durflinger, P.; Anderson, K.; Dezfulian, C.; Roberts, B.W.; Chansky, M.E.; Parrillo, J.E.; Hollenberg, S.M. Randomized Controlled trial of inhaled nitric oxide for the treatment of microcirculatory dysfunction in patients with sepsis*. Crit. Care Med. 2014, 42, 2482–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumbarton, T.C.; Maxan, A.; Farah, N.; Sharawy, N.; Zhou, J.; Nantais, J.; Lehmann, C. Tetrahydrobiopterin improves microcirculation in experimental sepsis. Clin. Hemorheol. Microcirc. 2017, 67, 15–24. [Google Scholar] [CrossRef] [PubMed]
- López, A.; Lorente, J.A.; Steingrub, J.; Bakker, J.; McLuckie, A.; Willatts, S.; Brockway, M.; Anzueto, A.; Holzapfel, L.; Breen, D.; et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: Effect on survival in patients with septic shock*. Crit. Care Med. 2004, 32, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Grover, R.; McLuckie, A.; Holzapfel, L.; Andersson, J.; Lodato, R.; Watson, D.; Grossman, S.; Donaldson, J.; Takala, J. Administration of the nitric oxide synthase inhibitor NG-methyl-l-arginine hydrochloride (546C88) by intravenous infusion for up to 72 hours can promote the resolution of shock in patients with severe sepsis: Results of a randomized, double-blind, placebo-controlled multicenter study (study no. 144-002)*. Crit. Care Med. 2004, 32, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.L.; Park, Y.S. Maintenance of cellular tetrahydrobiopterin homeostasis. BMB Rep. 2010, 43, 584–592. [Google Scholar] [CrossRef]
- Fredenburgh, L.E.; Perrella, M.A.; Barragan-Bradford, D.; Hess, D.R.; Peters, E.; Welty-Wolf, K.E.; Kraft, B.D.; Harris, R.S.; Maurer, R.; Nakahira, K.; et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS. JCI Insight 2018, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Nitric Oxide | Carbon Monoxide | Hydrogen Sulfide | |
|---|---|---|---|
| Formula and molecular weight | NO (30.01 g/mol) | CO (28.01 g/mol) | H2S (34.08 g/mol) |
| Biological half-life | Seconds | Several minutes | Seconds–minutes |
| Chemical reactivity | Very high | Moderate | Very high |
| Properties of free radicals | Yes | No | Yes |
| Endogenous production enzymes | nNOS; iNOS; eNOS; mtNOS a | HO-1; HO-2; HO-3 a | CBS; CSE; 3-MST/CAT; 3-MST/DAO b |
| Main substrates for biosynthesis | L-arginine | Heme | L-cysteine; 3-mercaptopyruvate; D-cysteine b |
| Clearance sources | Oxidization | Being exhaled from the airway; Binding to heme proteins; Oxidization c | Oxidization (mitochondrion); Methylation (cytoplasm); Being excreted from urine |
| End products | Nitrite and nitrate | Carboxyhemoglobin; Carbon dioxide c | Thiosulfate and sulfate; methanethiol |
| Second messenger | sGC | sGC | NA |
| Involvement in sepsis | Yes (Mainly detrimental) | Yes (Mainly beneficial) | Yes (Mainly detrimental) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.; Chambers, S.; Zeng, Y.; Bhatia, M. Gases in Sepsis: Novel Mediators and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 3669. https://doi.org/10.3390/ijms23073669
Zhu Z, Chambers S, Zeng Y, Bhatia M. Gases in Sepsis: Novel Mediators and Therapeutic Targets. International Journal of Molecular Sciences. 2022; 23(7):3669. https://doi.org/10.3390/ijms23073669
Chicago/Turabian StyleZhu, Zhixing, Stephen Chambers, Yiming Zeng, and Madhav Bhatia. 2022. "Gases in Sepsis: Novel Mediators and Therapeutic Targets" International Journal of Molecular Sciences 23, no. 7: 3669. https://doi.org/10.3390/ijms23073669