
Krüppel-like Transcription Factor 7 Is a Causal Gene in Autism Development


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
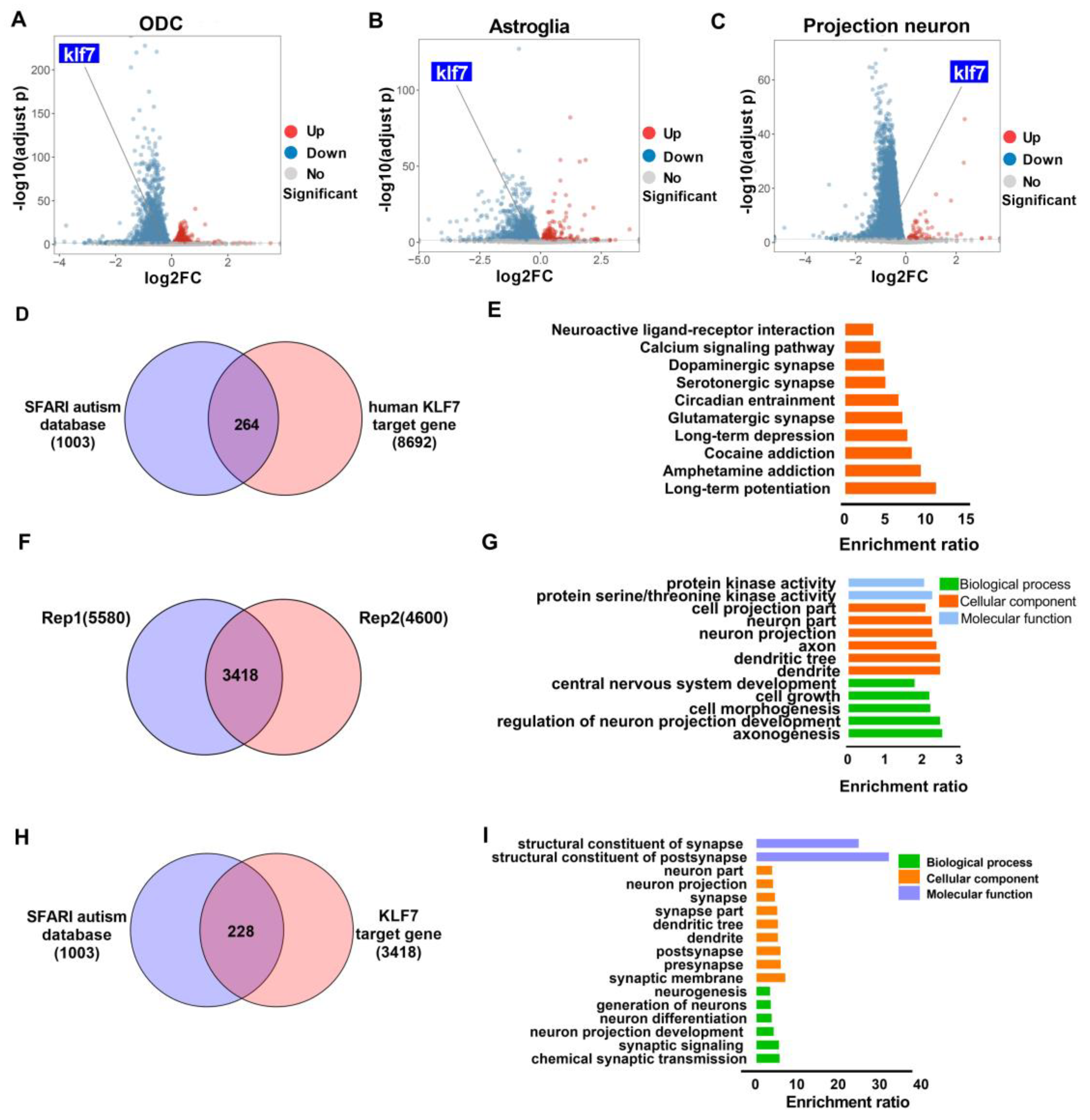
2.1. The Level of klf7 Is Altered in Human ASD Brain and klf7 Binds ASD Risk Genes
2.2. Effects of klf7 Deletion on the Adult Mouse
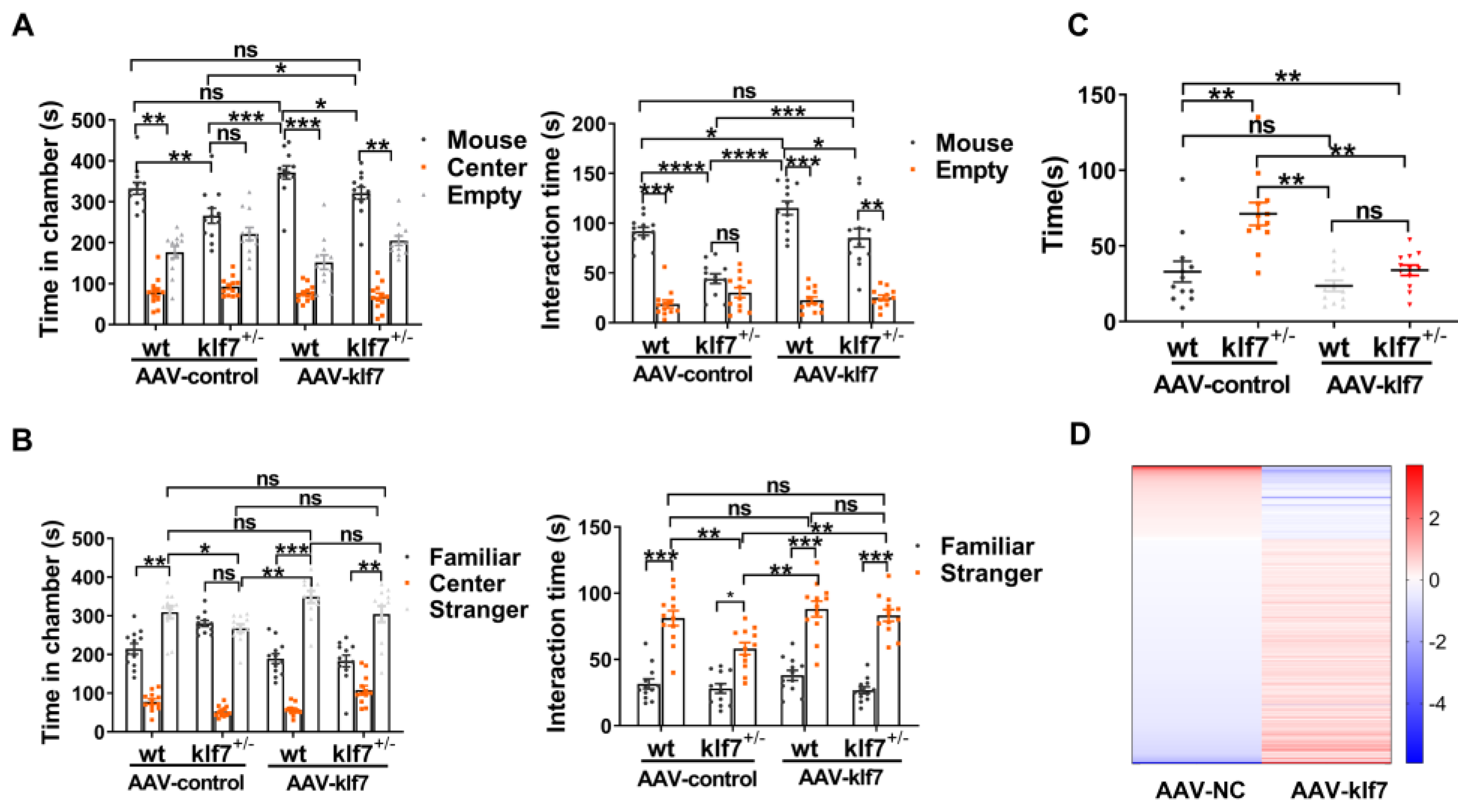
2.3. Klf7-Deficient Mice Show the Core Symptoms of ASD
2.4. Klf7-Deficient Mice Exhibit Other ASD-like Behaviors
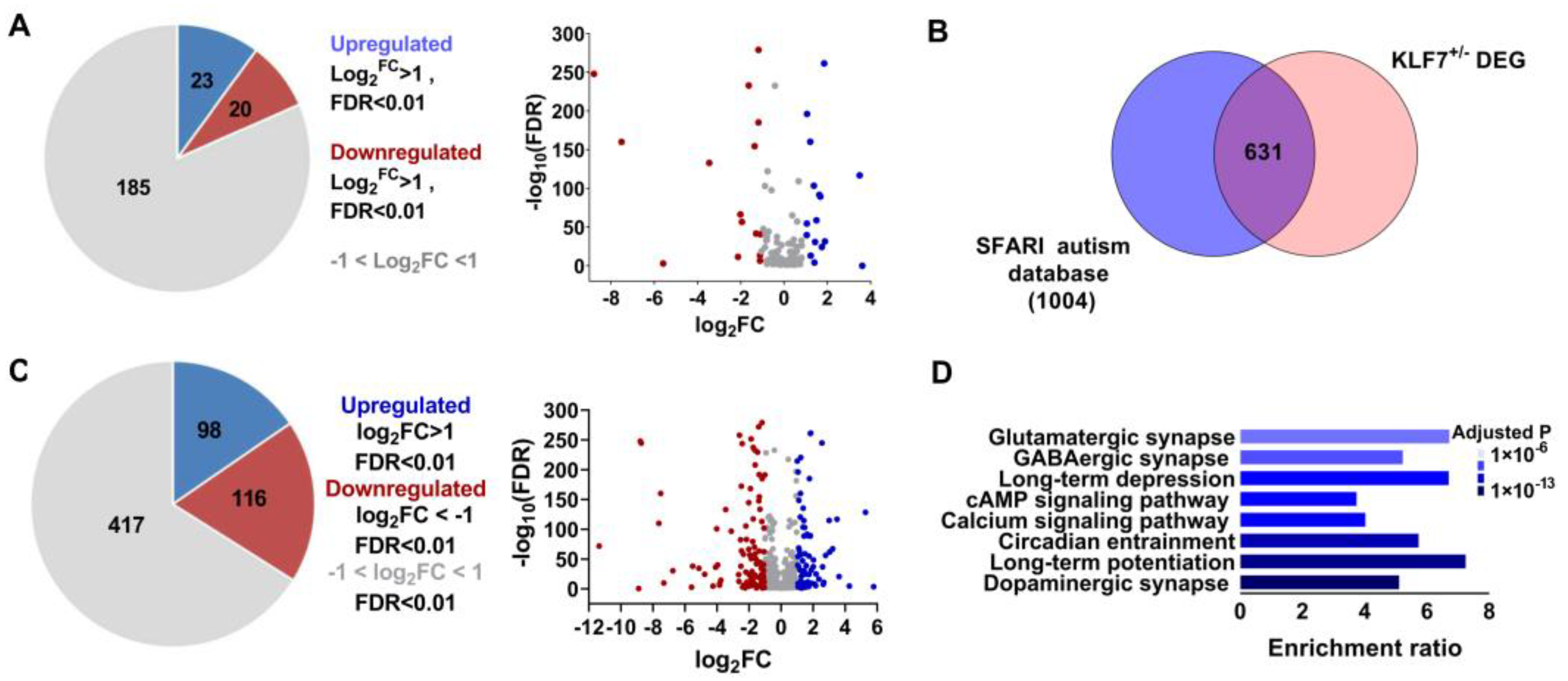
2.5. Klf7 Regulate a Large Number of Autism Genes
2.6. Increasing the Expression of klf7 Rescued the Core Symptom of ASD in klf7+/− Adult Mice
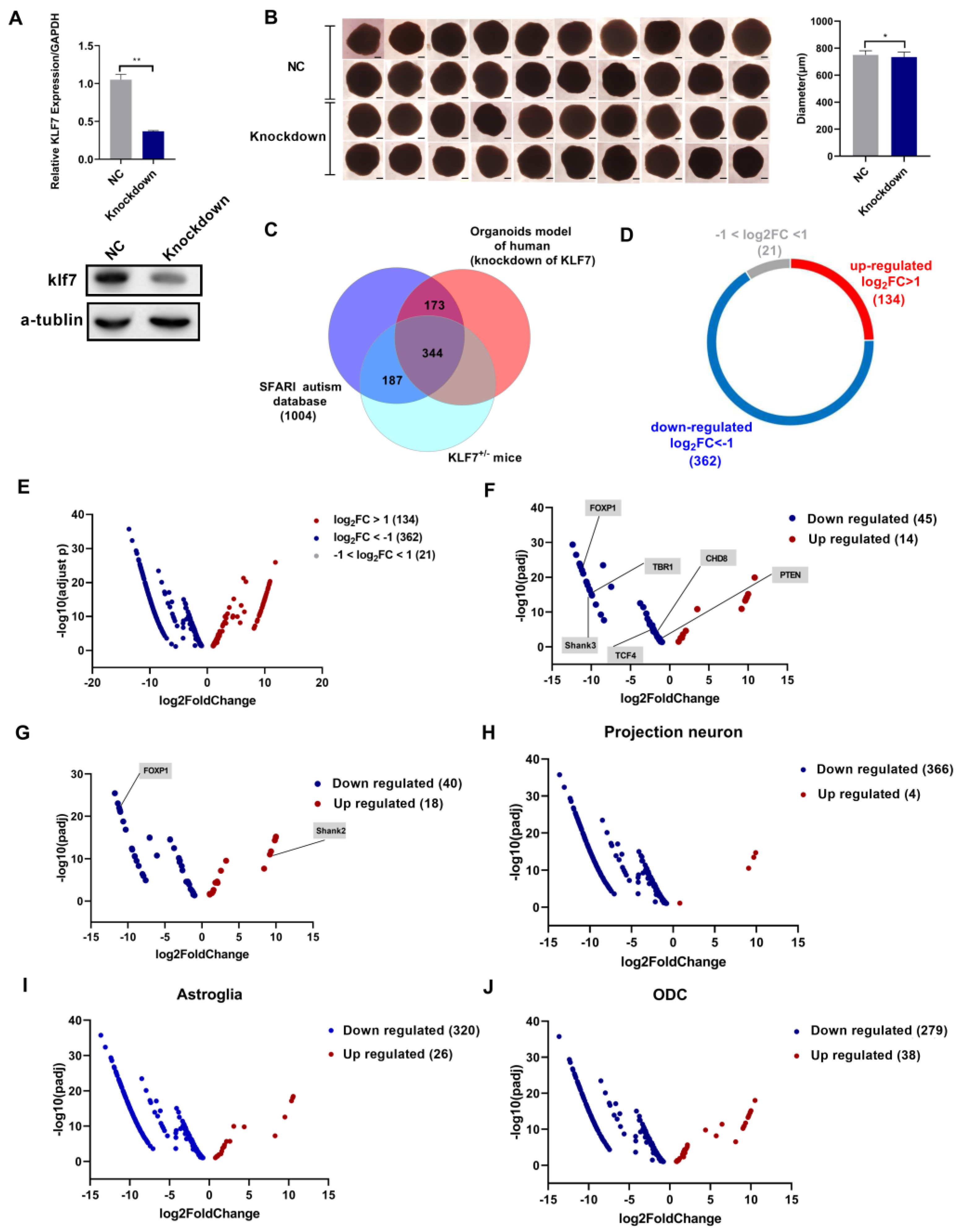
2.7. ASD Risk Genes Were Dysregulated in a klf7 Knockdown Human Brain Organoid Model
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Differential Gene Expression Analysis of Single-Cell Data from ASD Patient Brains
4.3. ChIP-Seq
4.4. Behavioral Tests
4.4.1. Olfactory Discrimination Test
4.4.2. Three-Chamber Test
4.4.3. Grooming
4.4.4. Novel Object Recognition Test
4.4.5. Y Maze Spontaneous Selection Experiment
4.4.6. Open Field Test
4.4.7. Morris Water Maze Test
4.4.8. Nest-Building Test
4.5. RNA-Seq and Analysis
4.6. Intravenous Administration
4.7. shRNA-Mediated klf7 Knockdown
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hyman, S.L.; Levy, S.E.; Myers, S.M.; Council On Children With Disabilities; Section on Developmental and Behavioral Pediatrics; Kuo, D.Z.; Apkon, S.; Davidson, L.F.; Ellerbeck, K.A.; Foster, J.E.A.; et al. Identification, Evaluation, and Management of Children With Autism Spectrum Disorder. Pediatrics 2020, 145, e20193447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, S.; Lu, Y.; Li, Y.; Shi, J.; Cui, H.; Gu, Y.; Li, Y.; Zhong, W.; Zhu, X.; Liu, Y.; et al. Prevalence of autism spectrum disorder in Asia: A systematic review and meta-analysis. Psychiatry Res. 2020, 284, 112679. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; Flint, J. Genetics and genomics of psychiatric disease. Science 2015, 349, 1489–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzzo, E.K.; Perez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocchetti, A.G.; Kopp, M.; Waltes, R.; Haslinger, D.; Duketis, E.; Jarczok, T.A.; Poustka, F.; Voran, A.; Graab, U.; Meyer, J.; et al. Variants of the CNTNAP2 5’ promoter as risk factors for autism spectrum disorders: A genetic and functional approach. Mol. Psychiatry 2015, 20, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef]
- Fakhro, K.A. Genomics of Autism. Adv. Neurobiol. 2020, 24, 83–96. [Google Scholar] [CrossRef]
- Fernandez, B.A.; Scherer, S.W. Syndromic autism spectrum disorders: Moving from a clinically defined to a molecularly defined approach. Dialogues Clin. Neurosci. 2017, 19, 353–371. [Google Scholar]
- Lin, G.N.; Corominas, R.; Lemmens, I.; Yang, X.; Tavernier, J.; Hill, D.E.; Vidal, M.; Sebat, J.; Iakoucheva, L.M. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron 2015, 85, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Iakoucheva, L.M.; Muotri, A.R.; Sebat, J. Getting to the Cores of Autism. Cell 2019, 178, 1287–1298. [Google Scholar] [CrossRef]
- Bieker, J.J. Kruppel-like factors: Three fingers in many pies. J. Biol Chem 2001, 276, 34355–34358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laub, F.; Dragomir, C.; Ramirez, F. Mice without transcription factor KLF7 provide new insight into olfactory bulb development. Brain Res. 2006, 1103, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, S.; Laub, F.; Else, C.; Dragomir, C.; Ramirez, F. Identification of MoKA, a novel F-box protein that modulates Kruppel-like transcription factor 7 activity. Mol. Cell Biol. 2004, 24, 1058–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.W.; Liu, X.H.; Zhao, Y.D.; Xiao, Z.; He, W.J.; Hu, Z.A.; Ruan, H.Z. Role of P2Y1 receptor in astroglia-to-neuron signaling at dorsal spinal cord. J. Neurosci. Res. 2009, 87, 2667–2676. [Google Scholar] [CrossRef]
- Sugiyama, S.; Yumimoto, K.; Inoue, I.; Nakayama, K.I. SCF(Fbxw7) ubiquitylates KLF7 for degradation in a manner dependent on GSK-3-mediated phosphorylation. Genes Cells 2019, 24, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Laub, F.; Lei, L.; Sumiyoshi, H.; Kajimura, D.; Dragomir, C.; Smaldone, S.; Puche, A.C.; Petros, T.J.; Mason, C.; Parada, L.F.; et al. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol. Cell Biol. 2005, 25, 5699–5711. [Google Scholar] [CrossRef] [Green Version]
- Brandau, D.T.; Lund, M.; Cooley, L.D.; Sanger, W.G.; Butler, M.G. Autistic and dysmorphic features associated with a submicroscopic 2q33.3-q34 interstitial deletion detected by array comparative genomic hybridization. Am. J. Med. Genet. A 2008, 146, 521–524. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Ballif, B.C.; Torchia, B.S.; Sahoo, T.; Ravnan, J.B.; Schultz, R.; Lamb, A.; Bejjani, B.A.; Shaffer, L.G. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet. Med. 2010, 12, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Jang, D.H.; Chae, H.; Kim, M. Autistic and Rett-like features associated with 2q33.3-q34 interstitial deletion. Am. J. Med. Genet. A 2015, 167, 2213–2218. [Google Scholar] [CrossRef]
- Courtens, W.; Speleman, F.; Messiaen, L.; Bormans, J.; Van Roy, N.; Vamos, E. Interstitial deletion 2q33.3-q34 in a boy with a phenotype resembling the Seckel syndrome. Am. J. Med. Genet. 1997, 71, 479–485. [Google Scholar] [CrossRef]
- Powis, Z.; Petrik, I.; Cohen, J.S.; Escolar, D.; Burton, J.; van Ravenswaaij-Arts, C.M.A.; Sival, D.A.; Stegmann, A.P.A.; Kleefstra, T.; Pfundt, R.; et al. De novo variants in KLF7 are a potential novel cause of developmental delay/intellectual disability, neuromuscular and psychiatric symptoms. Clin. Genet. 2018, 93, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Laub, F.; Aldabe, R.; Friedrich, V., Jr.; Ohnishi, S.; Yoshida, T.; Ramirez, F. Developmental expression of mouse Kruppel-like transcription factor KLF7 suggests a potential role in neurogenesis. Dev. Biol. 2001, 233, 305–318. [Google Scholar] [CrossRef] [Green Version]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Laub, F.; Lush, M.; Romero, M.; Zhou, J.; Luikart, B.; Klesse, L.; Ramirez, F.; Parada, L.F. The zinc finger transcription factor Klf7 is required for TrkA gene expression and development of nociceptive sensory neurons. Genes Dev. 2005, 19, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitges, F.W.; Radovani, E.; Najafabadi, H.S.; Barazandeh, M.; Campitelli, L.F.; Yin, Y.; Jolma, A.; Zhong, G.; Guo, H.; Kanagalingam, T.; et al. Multiparameter functional diversity of human C2H2 zinc finger proteins. Genome Res. 2016, 26, 1742–1752. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism. 2013, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Banerjee-Basu, S.; Packer, A. SFARI Gene: An evolving database for the autism research community. Dis. Model. Mech. 2010, 3, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Ilsley, M.D.; Gillinder, K.R.; Magor, G.W.; Huang, S.; Bailey, T.L.; Crossley, M.; Perkins, A.C. Kruppel-like factors compete for promoters and enhancers to fine-tune transcription. Nucleic. Acids Res. 2017, 45, 6572–6588. [Google Scholar] [CrossRef] [Green Version]
- Stockhorst, U.; Pietrowsky, R. Olfactory perception, communication, and the nose-to-brain pathway. Physiol. Behav. 2004, 83, 3–11. [Google Scholar] [CrossRef]
- Gomot, M.; Bernard, F.A.; Davis, M.H.; Belmonte, M.K.; Ashwin, C.; Bullmore, E.T.; Baron-Cohen, S. Change detection in children with autism: An auditory event-related fMRI study. Neuroimage 2006, 29, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Tan, N.; Zhu, X.; Yao, M.; Wang, Y.; Zhang, X.; Xu, Z. Sh3rf2 Haploinsufficiency Leads to Unilateral Neuronal Development Deficits and Autistic-Like Behaviors in Mice. Cell Rep. 2018, 25, 2963–2971.e2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, P.; Bouwknecht, J.A.; Teague, R.; Paylor, R.; Zoghbi, H.Y. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 2005, 14, 205–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e523. [Google Scholar] [CrossRef]
- Phelan, K.; McDermid, H.E. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2012, 2, 186–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [Green Version]
- Schoch, H.; Kreibich, A.S.; Ferri, S.L.; White, R.S.; Bohorquez, D.; Banerjee, A.; Port, R.G.; Dow, H.C.; Cordero, L.; Pallathra, A.A.; et al. Sociability Deficits and Altered Amygdala Circuits in Mice Lacking Pcdh10, an Autism Associated Gene. Biol. Psychiatry 2017, 81, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Tops, S.; Habel, U.; Radke, S. Genetic and epigenetic regulatory mechanisms of the oxytocin receptor gene (OXTR) and the (clinical) implications for social behavior. Horm. Behav. 2019, 108, 84–93. [Google Scholar] [CrossRef]
- Nishimori, K.; Takayanagi, Y.; Yoshida, M.; Kasahara, Y.; Young, L.J.; Kawamata, M. New aspects of oxytocin receptor function revealed by knockout mice: Sociosexual behaviour and control of energy balance. Prog. Brain Res. 2008, 170, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Caldwell, H.K.; Macbeth, A.H.; Tolu, S.G.; Young, W.S., 3rd. A conditional knockout mouse line of the oxytocin receptor. Endocrinology 2008, 149, 3256–3263. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Marchetto, M.C.; Bardy, C.; Gage, F.H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 2016, 17, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P. The rise of three-dimensional human brain cultures. Nature 2018, 553, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Yoon, S.J.; Tran, S.S.; Makinson, C.D.; Park, J.Y.; Andersen, J.; Valencia, A.M.; Horvath, S.; Xiao, X.; Huguenard, J.R.; et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021, 24, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, Y.; Nishiyama, M.; Shoji, H.; Ohkawa, Y.; Kawamura, A.; Sato, T.; Suyama, M.; Takumi, T.; Miyakawa, T.; Nakayama, K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016, 537, 675–679. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Parras, A.; Anta, H.; Santos-Galindo, M.; Swarup, V.; Elorza, A.; Nieto-Gonzalez, J.L.; Pico, S.; Hernandez, I.H.; Diaz-Hernandez, J.I.; Belloc, E.; et al. Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 2018, 560, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Wagnon, J.L.; Briese, M.; Sun, W.; Mahaffey, C.L.; Curk, T.; Rot, G.; Ule, J.; Frankel, W.N. CELF4 regulates translation and local abundance of a vast set of mRNAs, including genes associated with regulation of synaptic function. PLoS Genet. 2012, 8, e1003067. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.C.; Huang, T.N.; Hsueh, Y.P. T-Brain-1—A Potential Master Regulator in Autism Spectrum Disorders. Autism. Res. 2015, 8, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Cotney, J.; Muhle, R.A.; Sanders, S.J.; Liu, L.; Willsey, A.J.; Niu, W.; Liu, W.; Klei, L.; Lei, J.; Yin, J.; et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 2015, 6, 6404. [Google Scholar] [CrossRef]
- Canales, C.P.; Estes, M.L.; Cichewicz, K.; Angara, K.; Aboubechara, J.P.; Cameron, S.; Prendergast, K.; Su-Feher, L.; Zdilar, I.; Kreun, E.J.; et al. Sequential perturbations to mouse corticogenesis following in utero maternal immune activation. Elife 2021, 10, e60100. [Google Scholar] [CrossRef] [PubMed]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Baker, M.; Loree, J.; Kutanzi, K.; Hudson, D.; Pogribny, I.; Sedelnikova, O.; Bonner, W.; Kovalchuk, O. Epigenetic dysregulation underlies radiation-induced transgenerational genome instability in vivo. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Monteiro, P.; Zhou, Y.; Kim, J.A.; Gao, X.; Fu, Z.; Feng, G. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016, 530, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Sgritta, M.; Mays, J.; Zhou, H.; Lucero, R.; Park, J.; Wang, I.C.; Park, J.H.; Kaipparettu, B.A.; Stoica, L.; et al. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat. Med. 2019, 25, 1684–1690. [Google Scholar] [CrossRef]
- Penagarikano, O.; Lazaro, M.T.; Lu, X.H.; Gordon, A.; Dong, H.; Lam, H.A.; Peles, E.; Maidment, N.T.; Murphy, N.P.; Yang, X.W.; et al. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci. Transl. Med. 2015, 7, 271ra278. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, W.Y.; Jia, H.; Zhai, F.G.; Qu, W.R.; Cheng, Y.X.; Liu, Y.C.; Deng, L.X.; Guo, S.F.; Jin, Z.S. KLF7-transfected Schwann cell graft transplantation promotes sciatic nerve regeneration. Neuroscience 2017, 340, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Y.; Zhu, G.Y.; Yue, W.J.; Sun, G.D.; Zhu, X.F.; Wang, Y. KLF7 overexpression in bone marrow stromal stem cells graft transplantation promotes sciatic nerve regeneration. J. Neural. Eng. 2019, 16, 056011. [Google Scholar] [CrossRef] [PubMed]
- Melissa, P.S.W.; Phelim, Y.V.C.; Navaratnam, V.; Yoke Yin, C. DNA Microarray Analysis of Estrogen Responsive Genes in Ishikawa Cells by Glabridin. Biochem. Insights 2017, 10, 1178626417721676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odawara, J.; Harada, A.; Yoshimi, T.; Maehara, K.; Tachibana, T.; Okada, S.; Akashi, K.; Ohkawa, Y. The classification of mRNA expression levels by the phosphorylation state of RNAPII CTD based on a combined genome-wide approach. BMC Genomics 2011, 12, 516. [Google Scholar] [CrossRef] [Green Version]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef]
- Shoji, H.; Hagihara, H.; Takao, K.; Hattori, S.; Miyakawa, T. T-maze forced alternation and left-right discrimination tasks for assessing working and reference memory in mice. J. Vis. Exp. 2012, 60, e3300. [Google Scholar] [CrossRef]
- Deacon, R.M. Assessing nest building in mice. Nat. Protoc. 2006, 1, 1117–1119. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, H.; Qiao, S.; Zhao, Y.; Jin, X.; Wang, C.; Wang, R.; Wang, Y.; Jiao, Y.; Liu, Y.; Zhang, B.; et al. Krüppel-like Transcription Factor 7 Is a Causal Gene in Autism Development. Int. J. Mol. Sci. 2022, 23, 3376. https://doi.org/10.3390/ijms23063376
Tian H, Qiao S, Zhao Y, Jin X, Wang C, Wang R, Wang Y, Jiao Y, Liu Y, Zhang B, et al. Krüppel-like Transcription Factor 7 Is a Causal Gene in Autism Development. International Journal of Molecular Sciences. 2022; 23(6):3376. https://doi.org/10.3390/ijms23063376
Chicago/Turabian StyleTian, Hui, Shupei Qiao, Yufang Zhao, Xiyun Jin, Cao Wang, Ruiqi Wang, Yilin Wang, Yanwen Jiao, Ying Liu, Bosong Zhang, and et al. 2022. "Krüppel-like Transcription Factor 7 Is a Causal Gene in Autism Development" International Journal of Molecular Sciences 23, no. 6: 3376. https://doi.org/10.3390/ijms23063376