
Complete Models of p53 Better Inform the Impact of Hotspot Mutations

Abstract
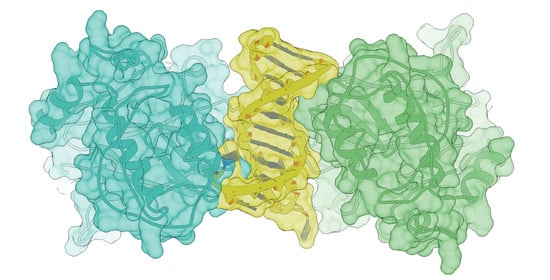
:
1. Introduction
2. Results
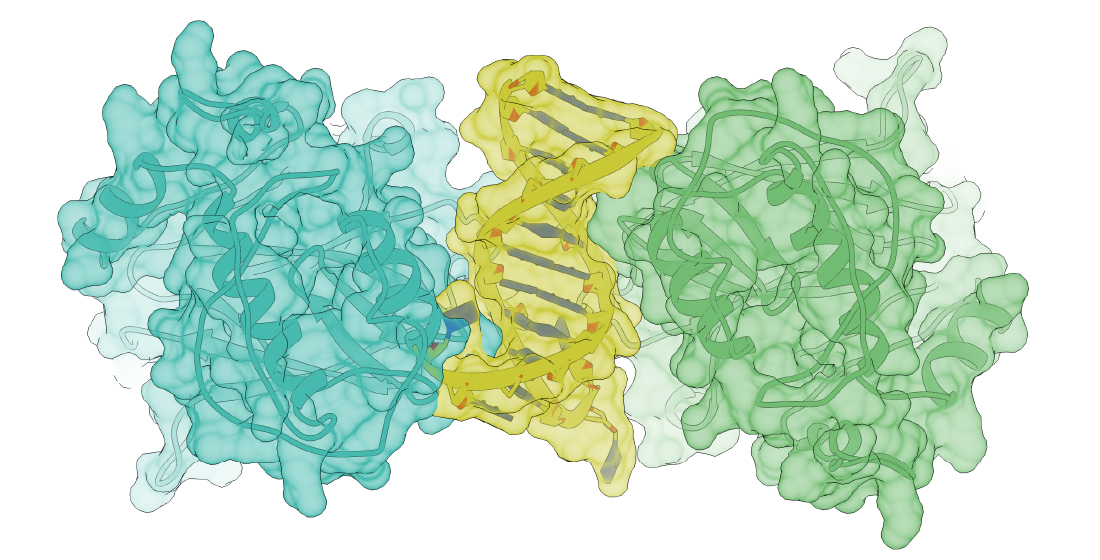
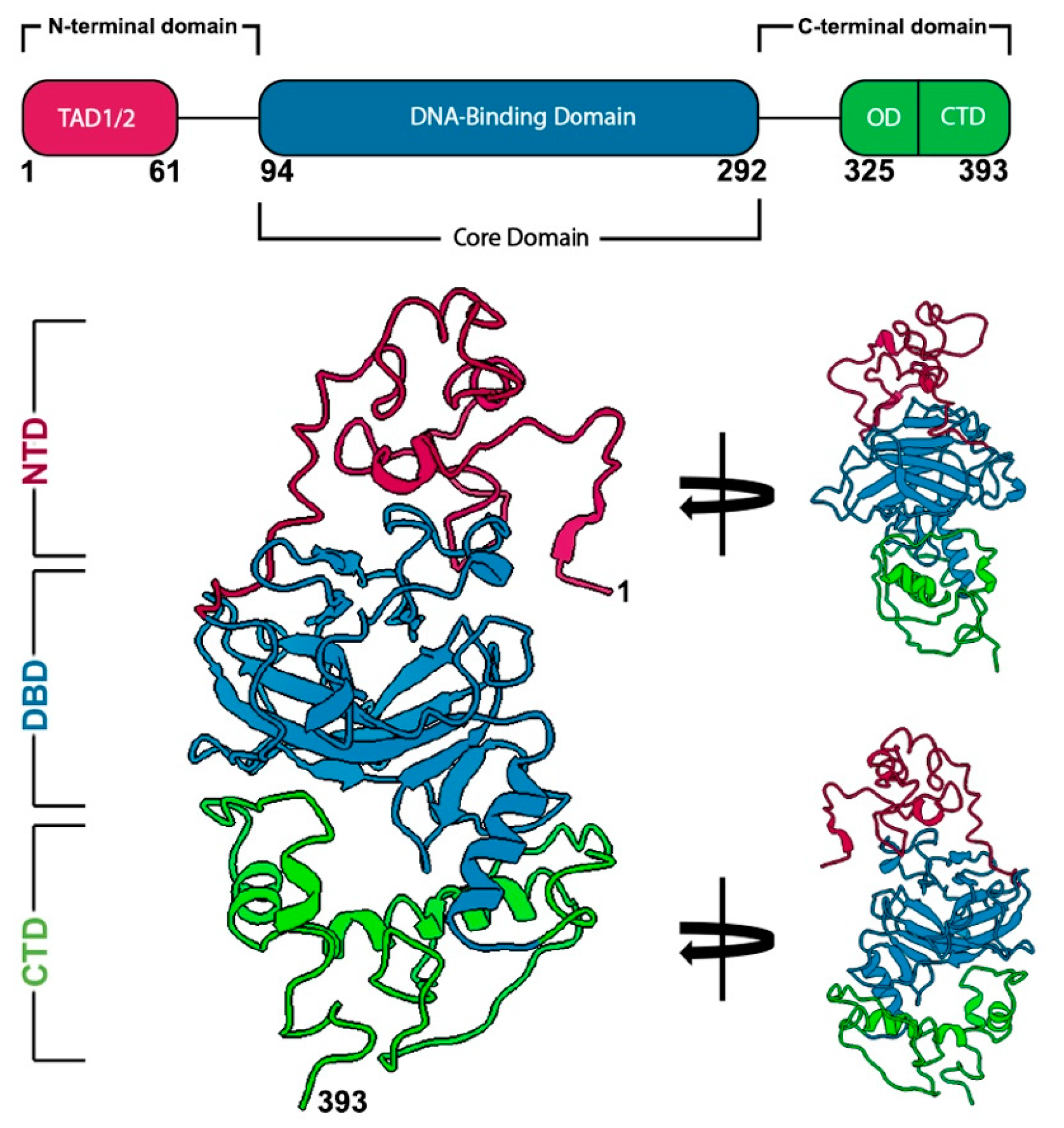
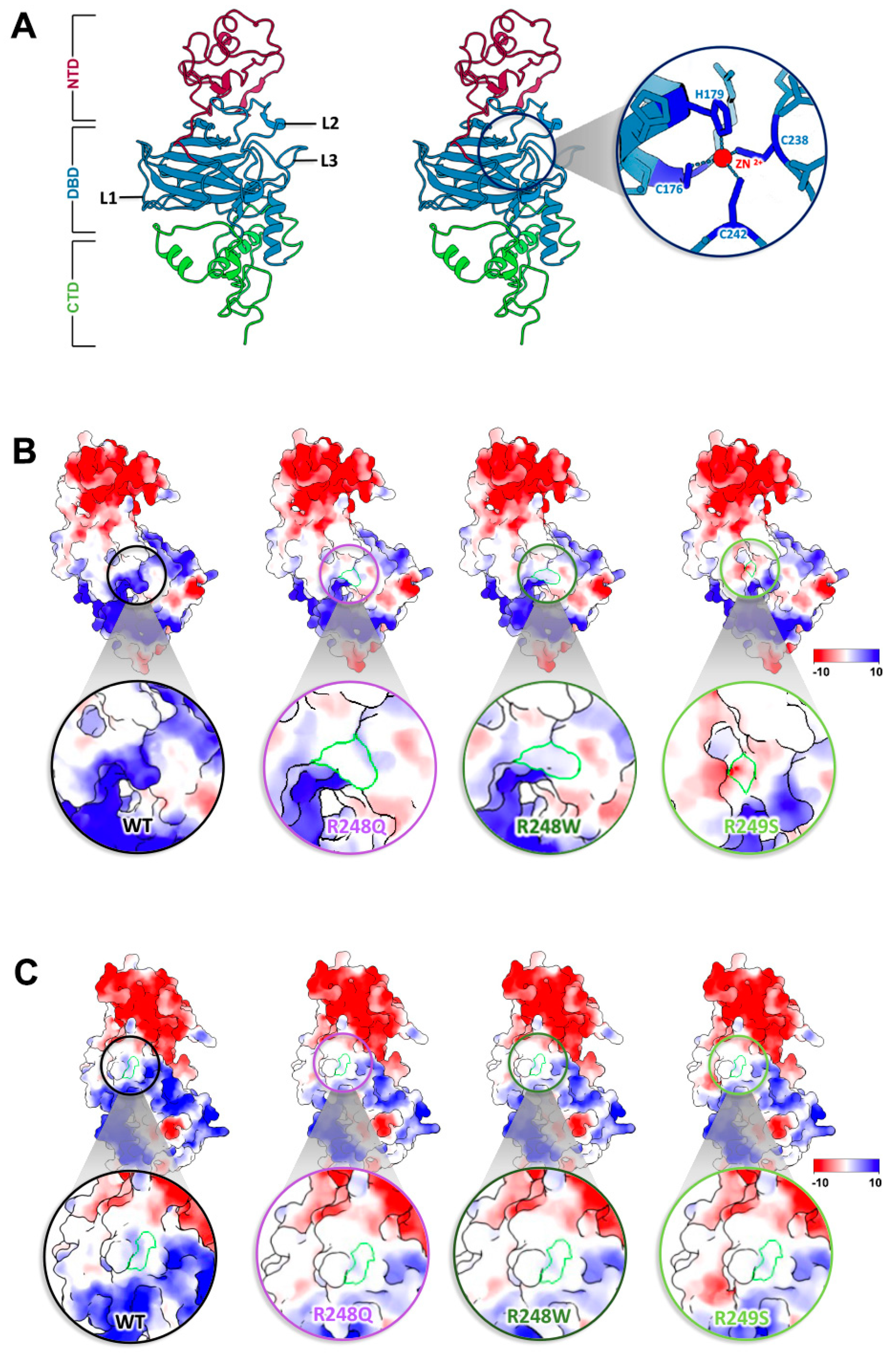
2.1. Discerning the Undiscernible: Producing p53 Models for Mutational Analysis
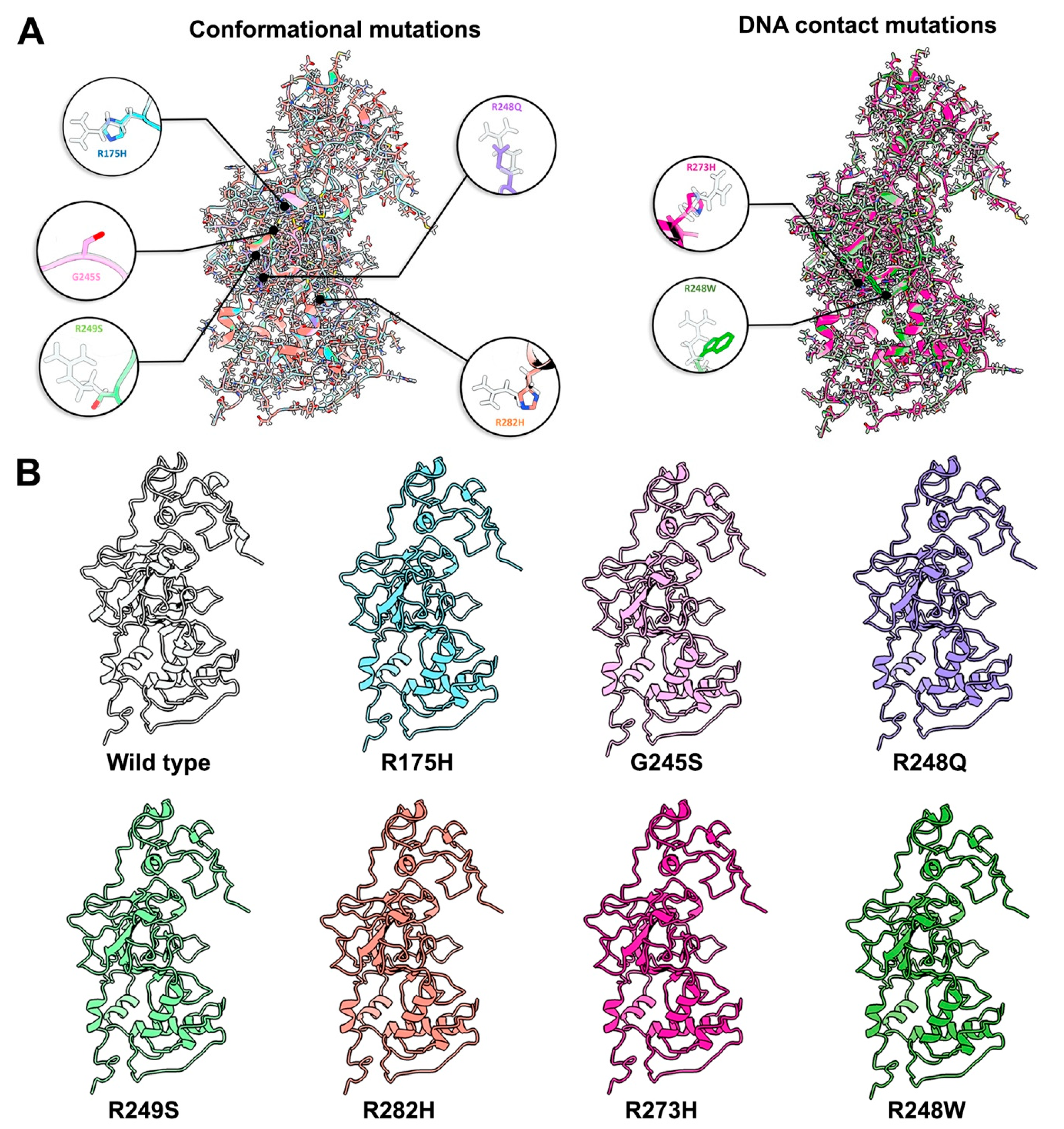
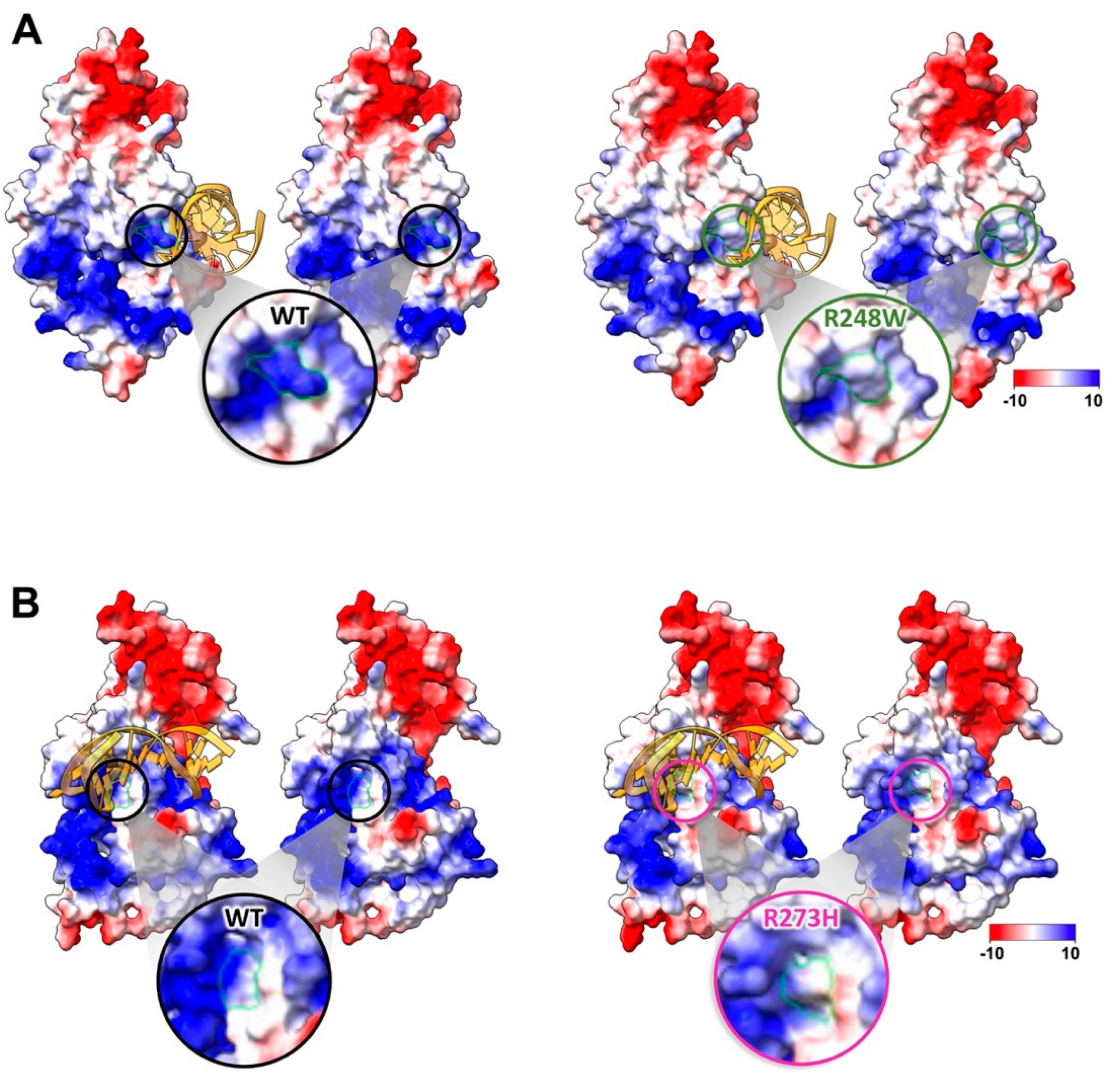
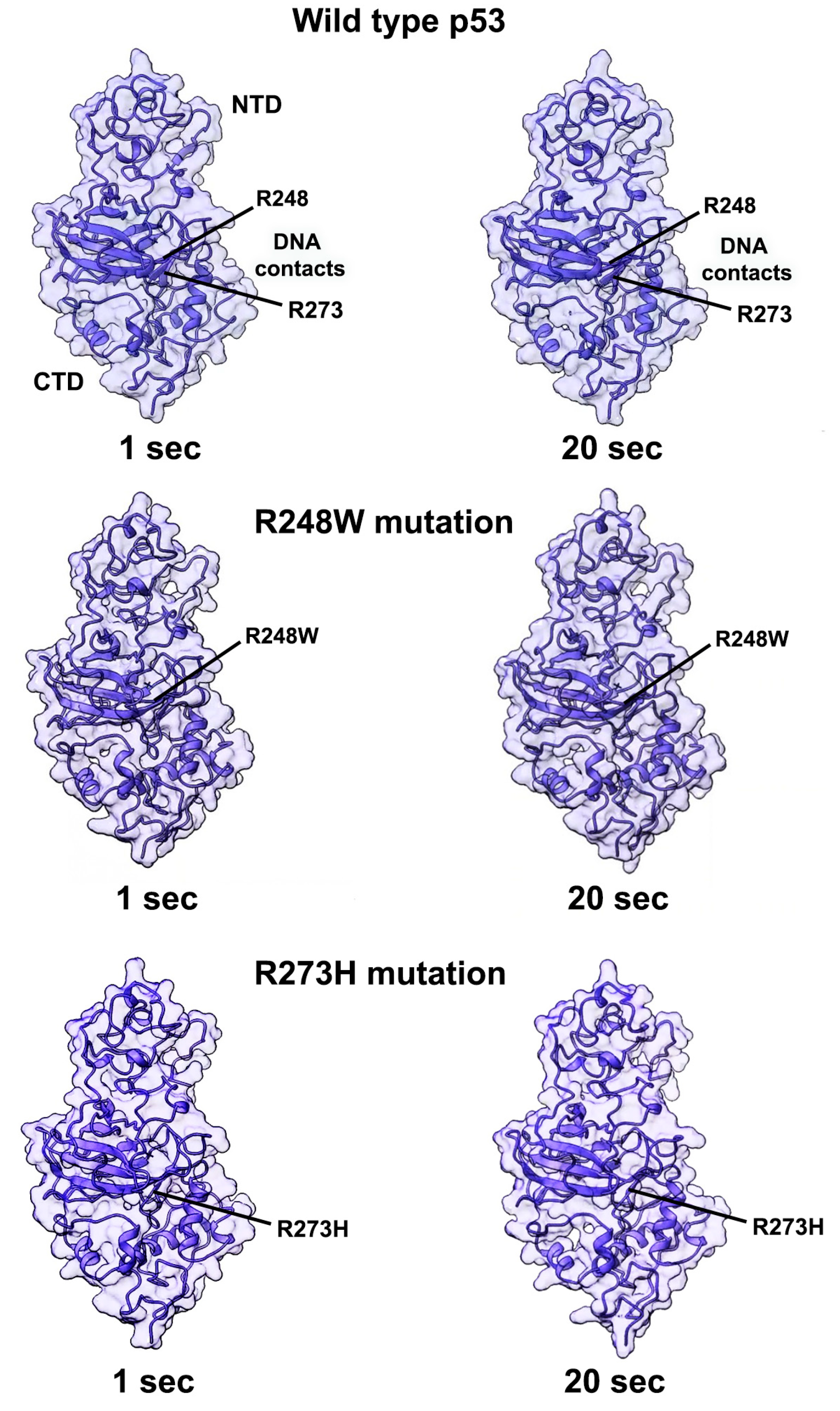
2.2. Conformational Mutations Have Rippling Structural Effects Due to Electrostatic Change
2.3. Contact Mutations Are Subtle but Detrimental to Overall p53 Function
3. Discussion
4. Methods and Materials
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vermund, H.; Gollin, F.F. Mechanisms of Action of Radiotherapy and Chemotherapeutic Adjuvants: A Review. Cancer 1968, 21, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Goodin, S. Oral Chemotherapeutic Agents: Understanding Mechanisms of Action and Drug Interactions. Am. J. Health Syst. Pharm. 2007, 64, S15–S24. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-E.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C. P53 Mutations in Human Cancers. Science 1991, 253, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lu, X. Live or Let Die: The Cell’s Response to P53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A Complex Barcode Underlies the Heterogeneous Response of P53 to Stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L. No PUMA, No Death. Cancer Cell 2003, 4, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, L. The Transcriptional Targets of P53 in Apoptosis Control. Biochem. Biophys. Res. Commun. 2005, 331, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. PUMA Cooperates with Direct Activator Proteins to Promote Mitochondrial Outer Membrane Permeabilization and Apoptosis. Cell Cycle 2009, 8, 2692–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hikisz, P.; Kiliańska, Z. Puma, a Critical Mediator of Cell Death—One Decade on from Its Discovery. Cell. Mol. Biol. Lett. 2012, 17, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Meyerhardt, J.A.; Mayer, R.J. Systemic Therapy for Colorectal Cancer. N. Engl. J. Med. 2005, 352, 476–487. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Mayer, R.J. Systemic Treatment of Colorectal Cancer. Gastroenterology 2008, 134, 1296–1310.e1. [Google Scholar] [CrossRef] [Green Version]
- Holch, J.; Stintzing, S.; Heinemann, V. Treatment of Metastatic Colorectal Cancer: Standard of Care and Future Perspectives. Visc. Med. 2016, 32, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Liu, N.; Liu, J.; Liu, Y.; Zhang, C.; Long, S.; Luo, G.; Zhang, L.; Zhang, Y. Mutant P53 Drives Cancer Chemotherapy Resistance Due to Loss of Function on Activating Transcription of PUMA. Cell Cycle 2019, 18, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The Tumor Suppressor P53: From Structures to Drug Discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [Green Version]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by P53 Tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef]
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA Recognition by P53 Revealed by Crystal Structures with Hoogsteen Base Pairs. Nat. Struct. Mol. Biol. 2010, 17, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Solares, M.J.; Jonaid, G.M.; Luqiu, W.Y.; Berry, S.; Khadela, J.; Liang, Y.; Evans, M.C.; Pridham, K.J.; Dearnaley, W.J.; Sheng, Z.; et al. High-Resolution Imaging of Human Cancer Proteins Using Microprocessor Materials. ChemBioChem 2022, 23, e202200310. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.R.; Bond, J.P.; Tarone, R.E.; Harris, C.C.; Makalowski, W.; Boguski, M.S.; Greenblatt, M.S. Evolutionary Conservation and Somatic Mutation Hotspot Maps of P53: Correlation with P53 Protein Structural and Functional Features. Oncogene 1999, 18, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepre, M.G.; Omar, S.I.; Grasso, G.; Morbiducci, U.; Deriu, M.A.; Tuszynski, J.A. Insights into the Effect of the G245S Single Point Mutation on the Structure of P53 and the Binding of the Protein to DNA. Molecules 2017, 22, 1358. [Google Scholar] [CrossRef] [Green Version]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic P53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 178–190.e8. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Gorina, S.; Jeffrey, P.; Pavletich, N. Crystal Structure of a P53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 1994, 265, 346. [Google Scholar] [CrossRef]
- Moore, P.S.; Sipos, B.; Orlandini, S.; Sorio, C.; Real, F.X.; Lemoine, N.R.; Gress, T.; Bassi, C.; Klöppel, G.; Kalthoff, H.; et al. Genetic Profile of 22 Pancreatic Carcinoma Cell Lines. Analysis of K-Ras, P53, P16 and DPC4/Smad4. Virchows Arch. 2001, 439, 798–802. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) Database and Website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- Liu, Y.; Bodmer, W.F. Analysis of P53 Mutations and Their Expression in 56 Colorectal Cancer Cell Lines. Proc. Natl. Acad. Sci. USA 2006, 103, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Markl, I.D.; Jones, P.A. Presence and Location of TP53 Mutation Determines Pattern of CDKN2A/ARF Pathway Inactivation in Bladder Cancer. Cancer Res. 1998, 58, 5348–5353. [Google Scholar] [PubMed]
- Gaidano, G.; Ballerini, P.; Gong, J.Z.; Inghirami, G.; Neri, A.; Newcomb, E.W.; Magrath, I.T.; Knowles, D.M.; Dalla-Favera, R. P53 Mutations in Human Lymphoid Malignancies: Association with Burkitt Lymphoma and Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 1991, 88, 5413–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Tainsky, M.A.; Bischoff, F.Z.; Strong, L.C.; Wahl, G.M. Wild-Type P53 Restores Cell Cycle Control and Inhibits Gene Amplification in Cells with Mutant P53 Alleles. Cell 1992, 70, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Iggo, R.; Gannon, J.; Lane, D.P. Genetic and Immunochemical Analysis of Mutant P53 in Human Breast Cancer Cell Lines. Oncogene 1990, 5, 893–899. [Google Scholar] [PubMed]
- Moneo, V.; Serelde, B.G.; Fominaya, J.; Leal, J.F.M.; Blanco-Aparicio, C.; Romero, L.; Sánchez-Beato, M.; Cigudosa, J.C.; Tercero, J.C.; Piris, M.A.; et al. Extreme Sensitivity to Yondelis (Trabectedin, ET-743) in Low Passaged Sarcoma Cell Lines Correlates with Mutated P53. J. Cell Biochem. 2007, 100, 339–348. [Google Scholar] [CrossRef]
- Tseng, J.E.; Rodriguez, M.; Ro, J.; Liu, D.; Hong, W.K.; Mao, L. Gender Differences in P53 Mutational Status in Small Cell Lung Cancer. Cancer Res. 1999, 59, 5666–5670. [Google Scholar]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural Basis for Understanding Oncogenic P53 Mutations and Designing Rescue Drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef] [Green Version]
- Zupnick, A.; Prives, C. Mutational Analysis of the P53 Core Domain L1 Loop. J. Biol. Chem. 2006, 281, 20464–20473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative Analysis of Residual Folding and DNA Binding in Mutant P53 Core Domain: Definition of Mutant States for Rescue in Cancer Therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.S.; Loh, S.N. Structure, Function, and Aggregation of the Zinc-Free Form of the P53 DNA Binding Domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef]
- Duan, J.; Nilsson, L. Effect of Zn 2+ on DNA Recognition and Stability of the P53 DNA-Binding Domain. Biochemistry 2006, 45, 7483–7492. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structure–Function–Rescue: The Diverse Nature of Common P53 Cancer Mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF CHIMERAX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Chen, H.; Sun, L.; Wang, M.; Wu, X.; Xiao, Z.-X.J. Hotspot Mutant P53-R273H Inhibits KLF6 Expression to Promote Cell Migration and Tumor Metastasis. Cell Death Dis. 2020, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. Ubiquitination, Phosphorylation and Acetylation: The Molecular Basis for P53 Regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Dai, C.; Gu, W. P53 Post-Translational Modification: Deregulated in Tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant P53 Gains Its Function via C-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol. Cell 2017, 68, 1134–1146.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, L.; Zhou, T.; Zhang, Z.; Zeng, C. Nicotine Promotes WRL68 Cells Proliferation Due to the Mutant P53 Gain-of-Function by Activating CDK6-P53-RS-PIN1-STAT1 Signaling Pathway. Chem. Res. Toxicol. 2020, 33, 2361–2373. [Google Scholar] [CrossRef]
- Wang, H.; Liao, P.; Zeng, S.X.; Lu, H. Co-Targeting P53-R249S and CDK4 Synergistically Suppresses Survival of Hepatocellular Carcinoma Cells. Cancer Biol. Ther. 2020, 21, 269–277. [Google Scholar] [CrossRef]
- Ku, B.M.; Kim, D.; Kim, K.; Yoo, B.C.; Kim, S.; Gong, Y.; Kim, S. Transglutaminase 2 Inhibition Found to Induce P53 Mediated Apoptosis in Renal Cell Carcinoma. FASEB J. 2013, 27, 3487–3495. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kang, J.H.; Ha, J.S.; Lee, J.-S.; Oh, S.-J.; Choi, H.-J.; Song, J.; Kim, S.-Y. Transglutaminase 2-Mediated P53 Depletion Promotes Angiogenesis by Increasing HIF-1α-P300 Binding in Renal Cell Carcinoma. IJMS 2020, 21, 5042. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting Mutant P53 for Cancer Therapy: Direct and Indirect Strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Sigal, A.; Rotter, V. Oncogenic Mutations of the P53 Tumor Suppressor: The Demons of the Guardian of the Genome. Cancer Res. 2000, 60, 6788–6793. [Google Scholar] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Croll, T.I. ISOLDE: A Physically Realistic Environment for Model Building into Low-Resolution Electron-Density Maps. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 519–530. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Impact on p53 | Topography | Cases Referenced |
|---|---|---|---|
| R175H | Conformational | Biliary tract, bones, brain, breast, colon, hematopoietic system, lung, ovary, pancreas, stomach urinary tract | [27,28,29,30] |
| G245S | Conformational | Brain, colon, colorectum, liver, pancreas, hematopoietic system, stomach | [28,29,30] |
| R248Q | Conformational | Bladder, bones, brain, breast, corpus uteri, esophagus, hematopoietic system, kidney, lung, lymph nodes | [28,30,31] |
| R248W | DNA contact | Brain, breast, colon, esophagus, hematopoietic system, kidney, lung, lymph nodes, skin | [28,30,32,33] |
| R249S | Conformational | Breast, colorectum, heart/medulla, liver, lung | [28,29,30,34] |
| R273H | DNA contact | Biliary tract, brain, colon/colorectum, corpus uteri, hematopoietic system, lung, lymph nodes, ovary, pancreas, skin, thyroid | [27,28,29,30,35] |
| R282H | Conformational | Lung | [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solares, M.J.; Kelly, D.F. Complete Models of p53 Better Inform the Impact of Hotspot Mutations. Int. J. Mol. Sci. 2022, 23, 15267. https://doi.org/10.3390/ijms232315267
Solares MJ, Kelly DF. Complete Models of p53 Better Inform the Impact of Hotspot Mutations. International Journal of Molecular Sciences. 2022; 23(23):15267. https://doi.org/10.3390/ijms232315267
Chicago/Turabian StyleSolares, Maria J., and Deborah F. Kelly. 2022. "Complete Models of p53 Better Inform the Impact of Hotspot Mutations" International Journal of Molecular Sciences 23, no. 23: 15267. https://doi.org/10.3390/ijms232315267