
Enhanced Acetaminophen Electrochemical Sensing Based on Nitrogen-Doped Graphene


Abstract
:1. Introduction
2. Results and Discussion
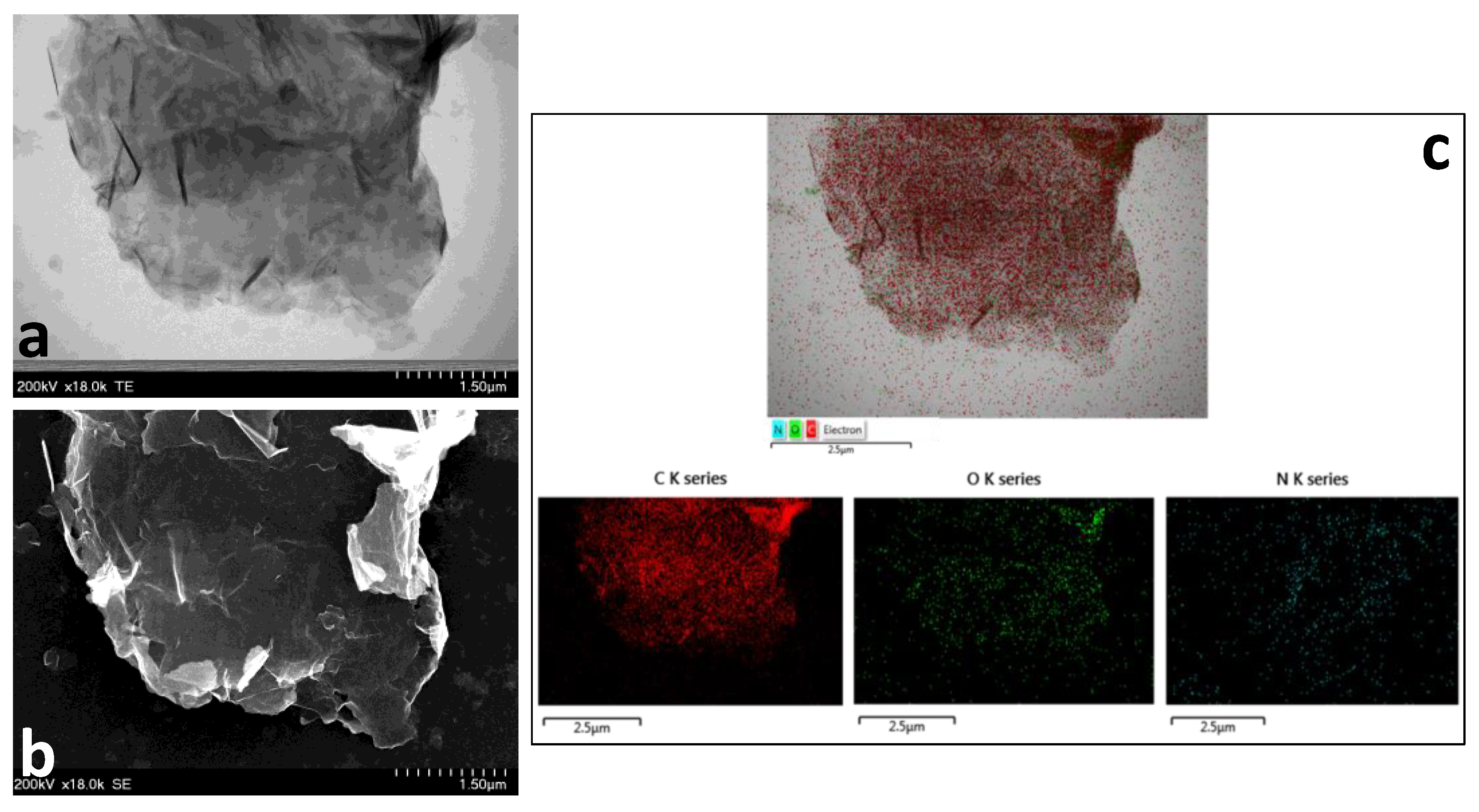
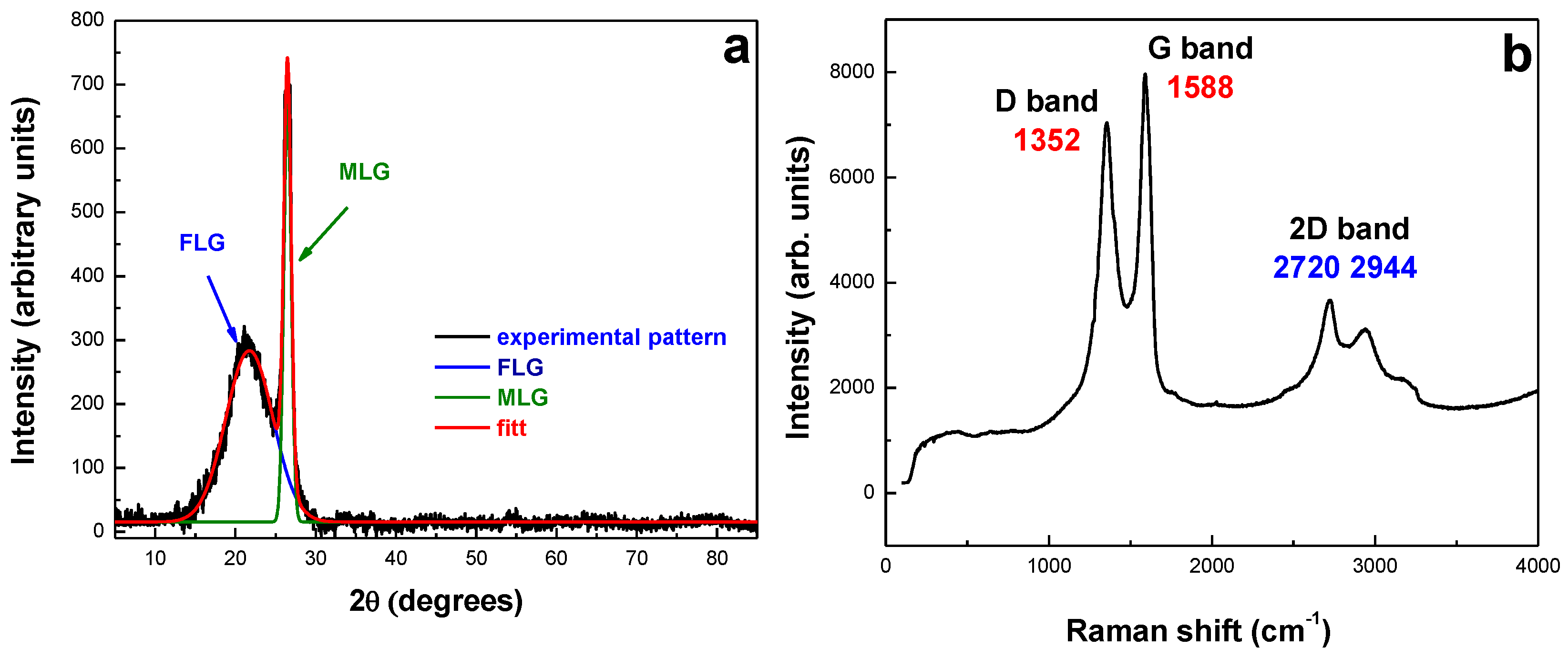
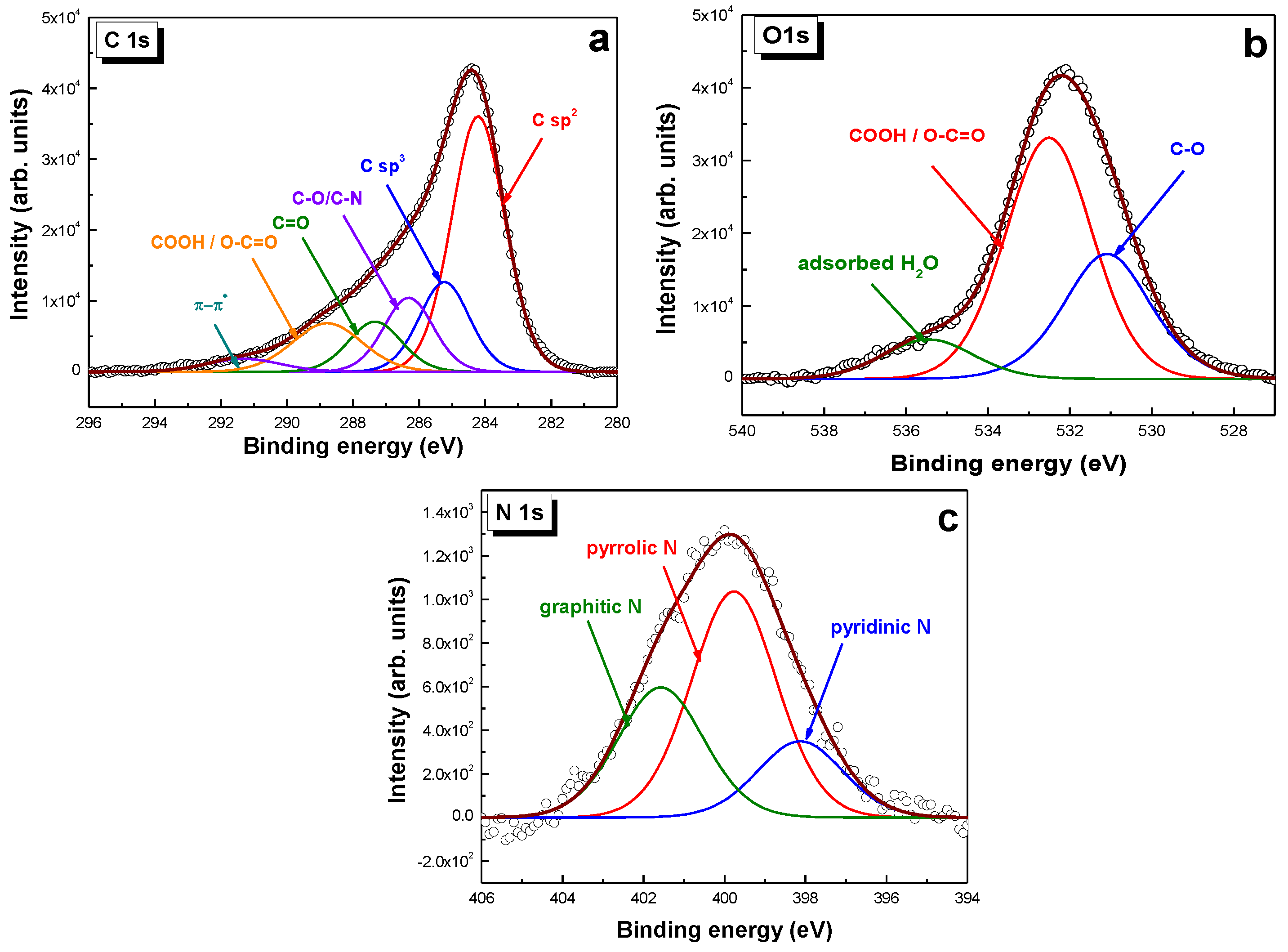
2.1. Morpho-Structural Material Characterization
2.2. Electrochemical Analysis
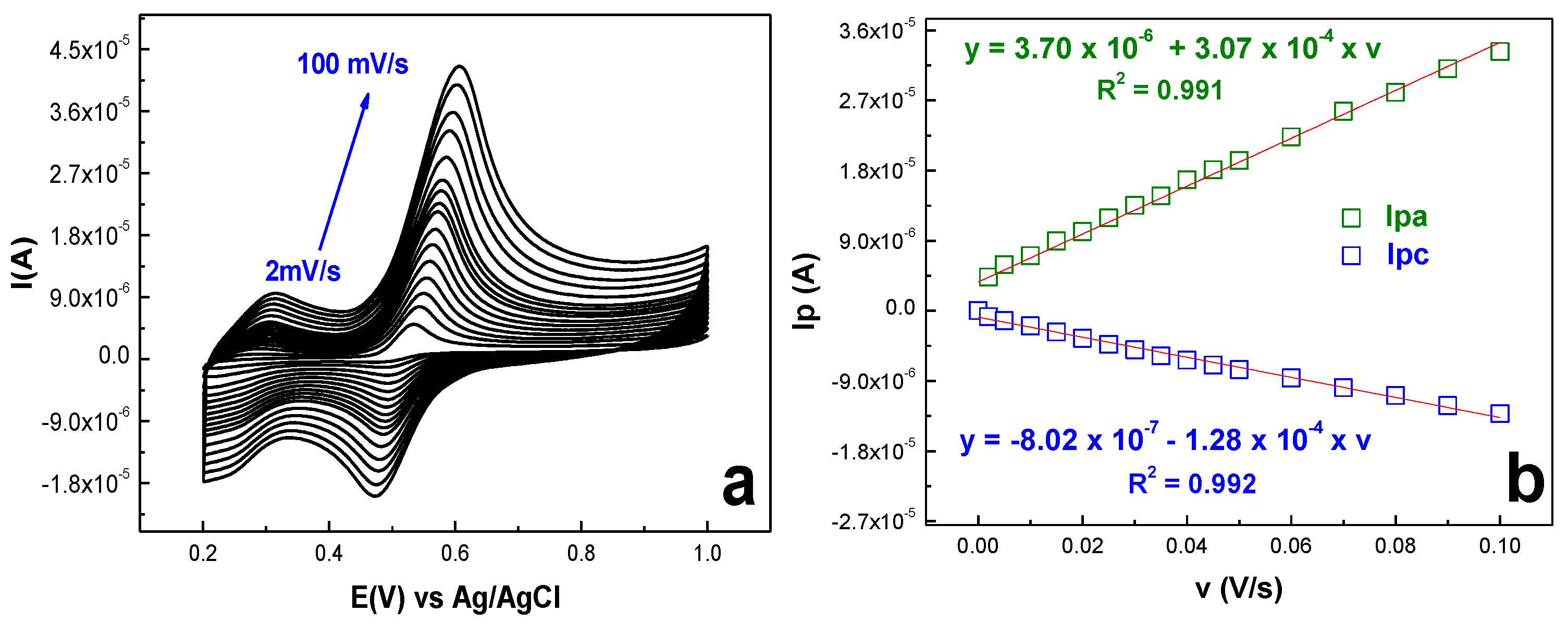
2.2.1. Modified Electrode Surface Area Determination
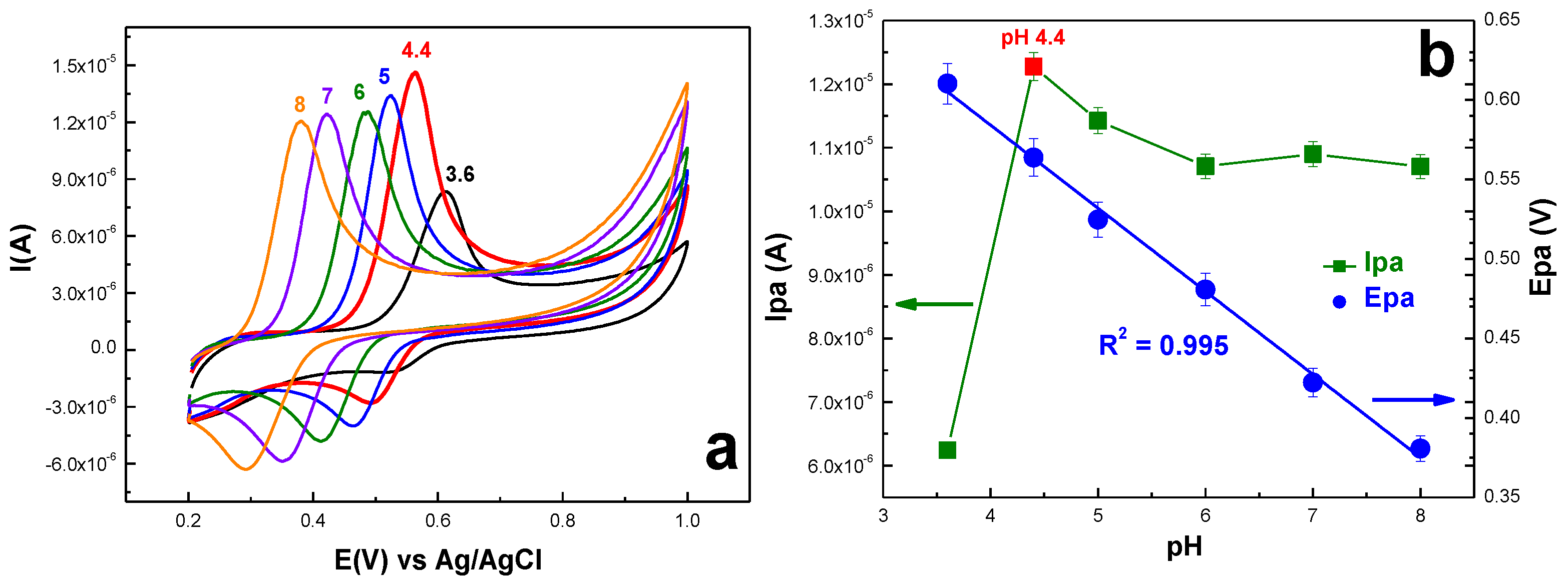
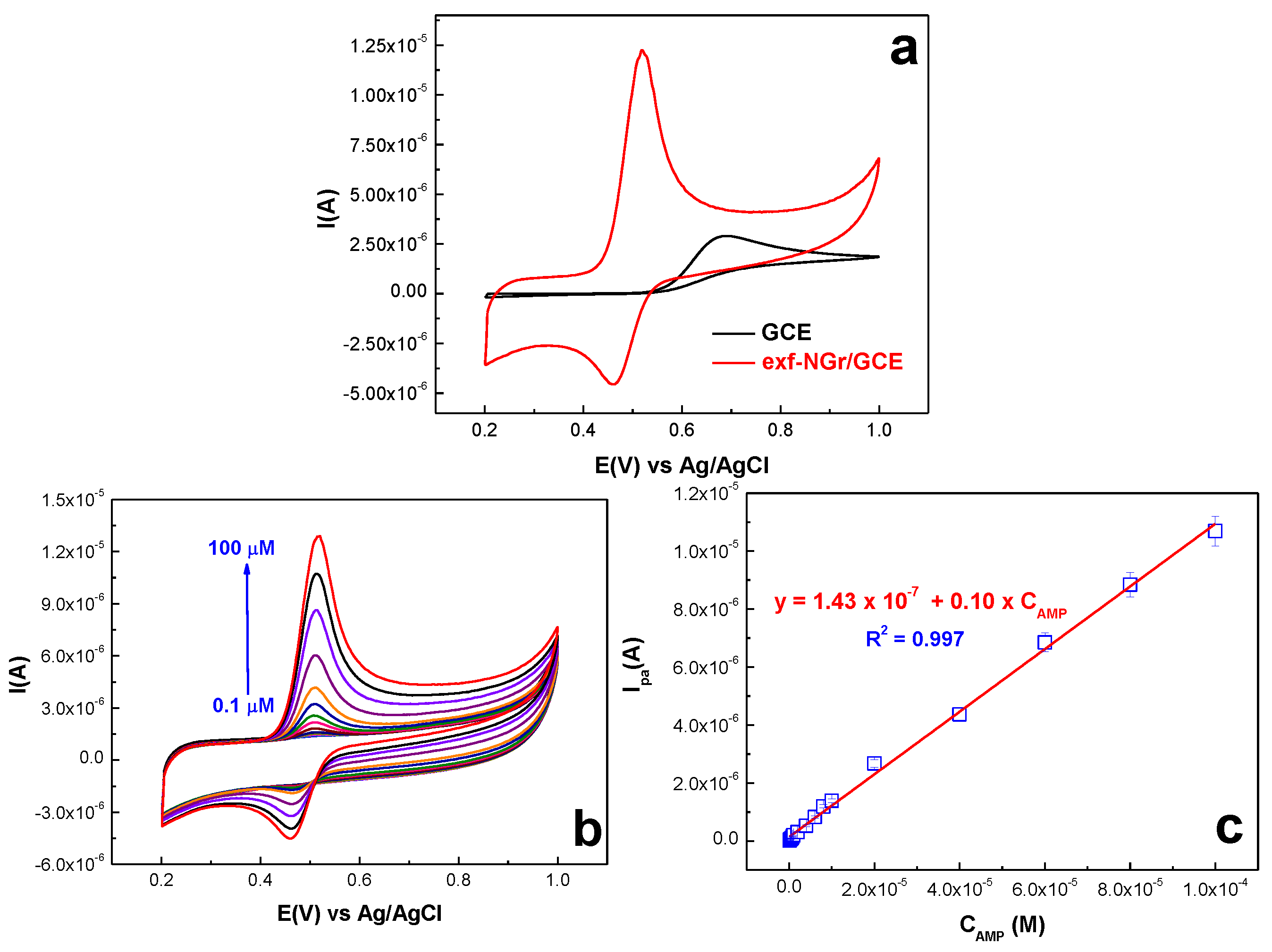
2.2.2. Acetaminophen Electrochemical Behavior at the Surface of exf-NGr/GCE
2.2.3. Calibration Curve and Limit of Detection
2.2.4. Reproducibility, Accuracy, Stability, and Interference Effects
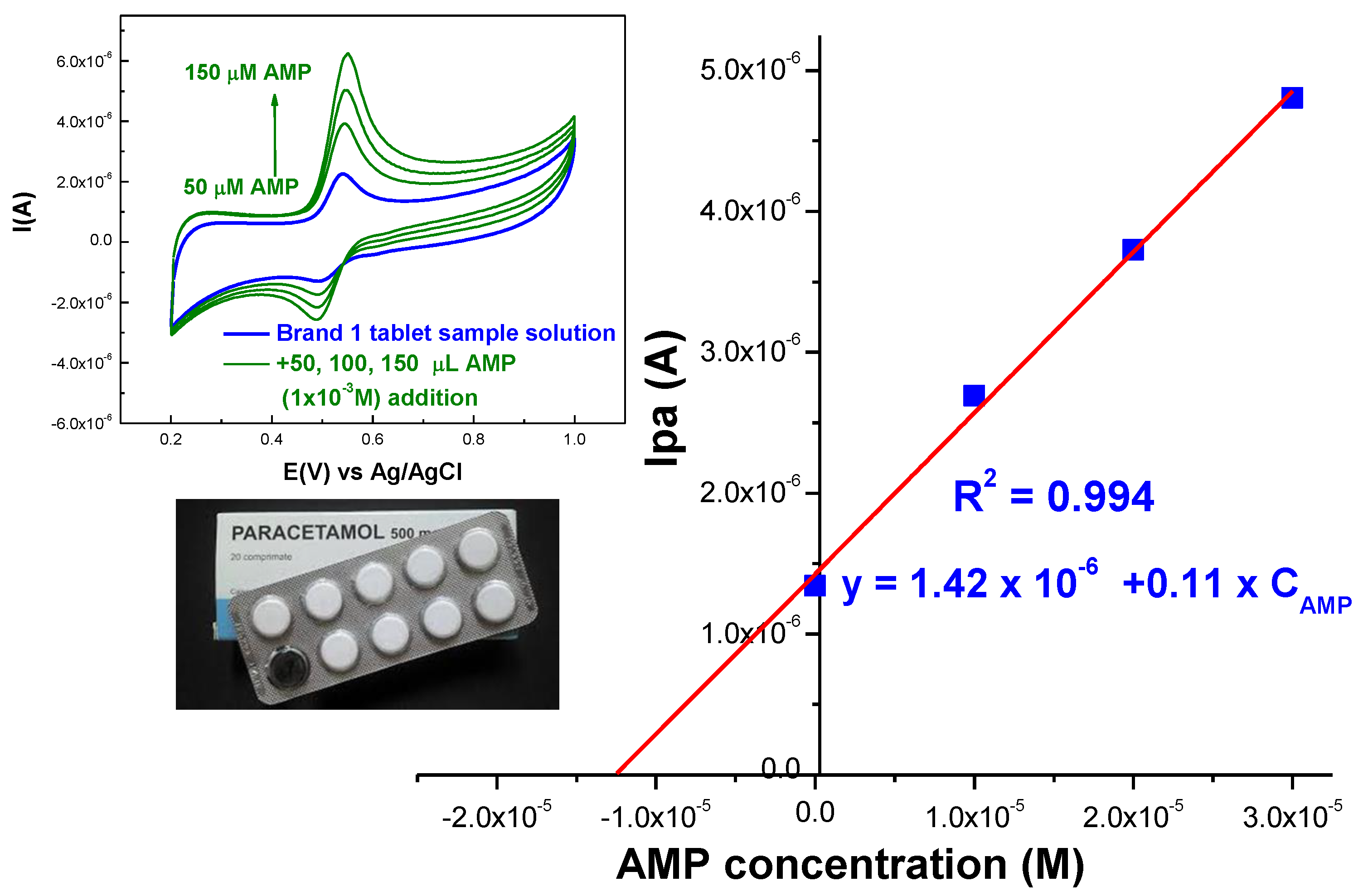
2.2.5. Real Sample Analysis
3. Materials and Methods
3.1. Reagents
3.2. Synthesis of Nitrogen-Doped Exfoliated Graphenes
3.3. Instruments
3.4. Electrochemical Experiments
3.4.1. Preparation of Nitrogen-Doped Graphene-Modified Electrodes (exf-NGr/GCE)
3.4.2. Electrochemical Setup
3.4.3. Preparation of Working Solutions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ennis, Z.N.; Dideriksen, D.; Vaegter, H.B.; Handberg, G.; Pottegard, A. Acetaminophen for Chronic Pain: A Systematic Review on Efficacy. Basic Clin. Pharmacol. Toxicol. 2016, 118, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannwarth, B.; Pehourcq, F. Pharmacologic basis for using paracetamol: Pharmacokinetic and pharmacodynamic issues. Drugs 2003, 63, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgman, M.J.; Garrard, A.R. A review of acetaminophen poisoning. Crit. Care Clin. 2012, 28, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Chiew, A.L.; Buckley, N.A. Acetaminophen Poisoning. Crit. Care Clin. 2021, 37, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Kostrubsky, S.E.; Sinclair, J.F.; Strom, S.C.; Wood, S.; Urda, E.; Stolz, D.B.; Wen, Y.H.; Kulkarni, S.; Mutlib, A. Phenobarbital and phenytoin increased acetaminophen hepatotoxicity due to inhibition of UDP-glucuronosyltransferases in cultured human hepatocytes. Toxicol. Sci. 2005, 87, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Perucca, E.; Richens, A. Paracetamol disposition in normal subjects and in patients treated with antiepileptic drugs. Br. J. Clin. Pharmacol. 1979, 7, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Minton, N.A.; Henry, J.A.; Frankel, R.J. Fatal paracetamol poisoning in an epileptic. Hum. Toxicol. 1988, 7, 33–34. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Q.; Liu, A.; Anadón, A.; Rodríguez, J.L.; Martínez-Larrañaga, M.R.; Yuan, Z.; Martínez, M.A. Paracetamol: Overdose-induced oxidative stress toxicity, metabolism, and protective effects of various compounds in vivo and in vitro. Drug Metab. Rev. 2017, 49, 395–437. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity-Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331. [Google Scholar] [CrossRef]
- Mitka, M. FDA asks physicians to stop prescribing high-dose acetaminophen products. JAMA 2014, 311, 563. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.G.; Letha, R. Determination of paracetamol in pure form and in dosage forms using N,N-dibromo dimethylhydantoin. J. Pharm. Biomed. Anal. 1997, 15, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Ahmad, S.; Singh, D.; Shukla, I.C. Titrimetric determination of dipyrone and paracetamol with potassium hexacyanoferrate(III) in an acidic medium. Analyst 1985, 110, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Llorent-Martínez, E.; Šatínský, D.; Solich, P.; Ortega-Barrales, P.; Molina-Díaz, A. Fluorimetric SIA optosensing in pharmaceutical analysis: Determination of paracetamol. J. Pharm. Biomed. Anal. 2007, 45, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, J.L.; Blanc, R.; Avidad, R.; Navalón, A. Spectrofluorimetric determination of paracetamol in pharmaceuticals and biological fluids. J. Pharm. Biomed. Anal. 1995, 13, 1119–1125. [Google Scholar] [CrossRef]
- Rele, R.V. Simultaneous UV-spectrophotometric estimation of paracetamol and guaiphenesin by first order derivative method in combined dosage form. Res. J. Pharm. Technol. 2016, 9, 509–512. [Google Scholar] [CrossRef]
- Glavanović, S.; Glavanović, M.; Tomišić, V. Simultaneous quantitative determination of paracetamol and tramadol in tablet formulation using UV spectrophotometry and chemometric methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2016, 157, 258–264. [Google Scholar] [CrossRef]
- Belal, T.; Awad, T.; Clark, R. Determination of paracetamol and tramadol hydrochloride in pharmaceutical mixture using HPLC and GC-MS. J. Chromatogr. Sci. 2009, 47, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Acheampong, A.; Gyasi, W.O.; Darko, G.; Apau, J.; Addai-Arhin, S. Validated RP-HPLC method for simultaneous determination and quantification of chlorpheniramine maleate, paracetamol and caffeine in tablet formulation. SpringerPlus 2016, 5, 625. [Google Scholar] [CrossRef] [Green Version]
- Farid, N.F.; Abdelaleem, E.A. HPTLC method for the determination of paracetamol, pseudoephedrine and loratidine in tablets and human plasma. J. Chromatogr. Sci. 2016, 54, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, C.; Wang, J.; Jiang, Y.; Fawcett, J.P.; Gu, J. Simultaneous quantitation of paracetamol, caffeine, pseudoephedrine, chlorpheniramine and cloperastine in human plasma by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010, 51, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Emre, D.; Özaltın, N. Simultaneous determination of paracetamol, caffeine and propyphenazone in ternary mixtures by micellar electrokinetic capillary chromatography. J. Chromatogr. B Biomed. Appl. 2007, 847, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Ruengsitagoon, W.; Liawruangrath, S.; Townshend, A. Flow injection chemiluminescence determination of paracetamol. Talanta 2006, 69, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Zidan, M.; Tee, T.W.; Abdullah, A.H.; Zainal, Z.; Kheng, G.J. Electrochemical oxidation of paracetamol mediated by nanoparticles bismuth oxide modified glassy carbon electrode. Int. J. Electrochem. Sci. 2011, 6, 279–288. [Google Scholar]
- Luo, J.; Fan, C.; Wang, X.; Liu, R.; Liu, X. A novel electrochemical sensor for paracetamol based on molecularly imprinted polymeric micelles. Sens. Actuators B Chem. 2013, 188, 909–916. [Google Scholar] [CrossRef]
- Özcan, L.; Şahin, Y. Determination of paracetamol based on electropolymerized-molecularly imprinted polypyrrole modified pencil graphite electrode. Sens. Actuators B Chem. 2007, 127, 362–369. [Google Scholar] [CrossRef]
- Thomas, T.; Mascarenhas, R.J.; Cotta, F.; Guha, K.S.; Swamy, B.E.; Martis, P.; Mekhalif, Z. Poly(Patton and Reeder’s reagent) modified carbon paste electrode for the sensitive detection of acetaminophen in biological fluid and pharmaceutical formulations. Colloids Surf. B Biointerfaces 2013, 101, 91–96. [Google Scholar] [CrossRef]
- Charithra, M.M.; Manjunatha, J.G. Enhanced voltammetric detection of Paracetamol by using carbon nanotube modified electrode as an electrochemical sensor. J. Electrochem. Sci. Eng. 2020, 10, 29–40. [Google Scholar] [CrossRef]
- Pushpanjali, P.A.; Manjunatha, J.G.; Hareesha, N.; D’Souza, E.S.; Charithra, M.M.; Prinith, N.S. Voltammetric analysis of antihistamine drug cetirizine and paracetamol at poly (L-Leucine) layered carbon nanotube paste electrode. Surface. Inter. 2021, 24, 101154. [Google Scholar] [CrossRef]
- Alam, A.U.; Qin, Y.; Howlader, M.M.R.; Hu, N.-X.; Deen, M.J. Electrochemical sensing of acetaminophen using multi-walled carbon nanotube and β-cyclodextrin. Sens. Actuators B Chem. 2018, 254, 896–909. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, J.H.; Lu, H.T.; Zhang, Q. Electrochemical behaviour and voltammetric determination of paracetamol on Nafion/TiO2-graphene modified glassy carbon electrode. Colloids Surf. B Biointerfaces 2011, 85, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sánchez, C.; Montiel-González, F.; Rodríguez-González, V. Electrochemical sensing of acetaminophen using a practical carbon paste electrode modified with a graphene oxide-Y2O3 nanocomposite. J. Taiwan Inst. Chem. Eng. 2019, 96, 382–389. [Google Scholar] [CrossRef]
- Qu, L.; Liu, Y.; Baek, J.B.; Dai, L. Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells. ACS Nano 2010, 4, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ma, L.; Jin, Z. Nitrogen-doped graphene: Synthesis, characterizations and energy applications. J. Energy Chem. 2018, 27, 146–160. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shi, W.; Zhang, R.; Luan, C.; Zeng, Q.; Wang, C.; Li, S.; Huang, Z.; Liao, H.; Ji, X. Electrochemical exfoliation of graphite into nitrogen-doped graphene in glycine solution and its energy storage properties. Electrochim. Acta 2016, 204, 100–107. [Google Scholar] [CrossRef]
- Wei, D.H.; Liu, Y.Q.; Wang, Y.; Zhang, H.L.; Huang, L.P.; Yu, G. Synthesis of N-Doped Graphene by Chemical Vapor Deposition and Its Electrical Properties. Nano Lett. 2009, 9, 1752–1758. [Google Scholar] [CrossRef]
- Li, M.; Wu, Z.S.; Ren, W.C.; Cheng, H.M.; Tang, N.J.; Wu, W.B.; Zhong, W.; Du, Y.W. The doping of reduced graphene oxide with nitrogen and its effect on the quenching of the material’s photoluminescence. Carbon 2012, 50, 5286–5291. [Google Scholar] [CrossRef]
- Lin, Z.Y.; Waller, G.H.; Liu, Y.; Liu, M.L.; Wong, C.P. 3D Nitrogen-doped graphene prepared by pyrolysis of graphene oxide with polypyrrole for electrocatalysis of oxygen reduction reaction. Nano Energy 2013, 2, 241–248. [Google Scholar] [CrossRef]
- Guan, L.; Cui, L.; Lin, K.; Wang, Y.Y.; Wang, X.T.; Jin, F.M.; He, F.; Chen, X.P.; Cui, S. Preparation of few-layer nitrogen-doped graphene nanosheets by DC arc discharge under nitrogen atmosphere of high temperature. Appl. Phys. A 2011, 102, 289–294. [Google Scholar] [CrossRef]
- Soin, N.; Roy, S.S.; Roy, S.; Hazra, K.S.; Misra, D.S.; Lim, T.H.; Hetherington, C.J.; Malaughlin, J.A. Enhanced and stable field emission from in situ nitrogen-doped few-layered graphene nanoflakes. J. Phys. Chem. C 2011, 115, 5366–5372. [Google Scholar] [CrossRef]
- Sun, L.; Wang, L.; Tian, C.G.; Tan, T.X.; Xie, Y.; Shi, K.Y.; Li, M.T.; Fu, H.G. Nitrogen-doped graphene with high nitrogen level via a one-step hydrothermal reaction of graphene oxide with urea for superior capacitive energy storage. RSC Adv. 2012, 2, 4498–4506. [Google Scholar] [CrossRef]
- Choi, C.H.; Park, S.H.; Chung, M.W.; Woo, S.I. Easy and controlled synthesis of nitrogen-doped carbon. Carbon 2013, 55, 98–107. [Google Scholar] [CrossRef]
- Xin, Y.C.; Liu, J.G.; Jie, X.; Liu, W.M.; Liu, F.Q.; Yin, Y.; Gu, J.; Zou, Z.G. Preparation and electrochemical characterization of nitrogen doped graphene by microwave as supporting materials for fuel cell catalysts. Electrochim. Acta 2012, 60, 354–358. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Fugane, K.; Mori, T.; Niu, L.; Ye, J.H. Wet chemical synthesis of nitrogen-doped graphene towards oxygen reduction electrocatalysts without high-temperature pyrolysis. J. Mater. Chem. 2012, 22, 6575–6580. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Cao, B.; Zhang, B.; Qi, X.; Pan, C.X. The production of nitrogen-doped graphene from mixed amine plus ethanol flames. Thin Solid Films 2012, 520, 6850–6855. [Google Scholar] [CrossRef]
- Zhang, L.; Xia, Z. Mechanisms of Oxygen Reduction Reaction on Nitrogen-Doped Graphene for Fuel Cells. J. Phys. Chem. C 2011, 115, 11170–11176. [Google Scholar] [CrossRef]
- Warren, B.E. X-ray Diffraction; Addison-Wesley: Reading, MA, USA, 1990; ISBN 0-486-66317-5. [Google Scholar]
- Pope, C.G. X-Ray Diffraction and the Bragg Equation. J. Chem. Educ. 1997, 74, 129. [Google Scholar] [CrossRef]
- Botas, C.; Álvarez, P.; Blanco, C.; Santamaría, R.; Granda, M.; Gutiérrez, M.D.; Rodríguez-Reinoso, F.; Menéndez, R. Critical temperatures in the synthesis of graphene-like materials by thermal exfoliation—Reduction of graphite oxide. Carbon 2013, 52, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sun, G.; Routh, P.; Kim, D.-H.; Huang, W.; Chen, P. Heteroatom-doped graphene materials: Syntheses, properties and applications. Chem. Soc. Rev. 2014, 43, 7067–7098. [Google Scholar] [CrossRef] [Green Version]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. 2009, 473, 51–87. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 2006, 97, 187401. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.S.; White, W.B. Characterization of diamond films by Raman spectroscopy. J. Mater. Res. 1989, 4, 385–393. [Google Scholar] [CrossRef]
- Magerusan, L.; Pogacean, F.; Coros, M.; Socaci, C.; Pruneanu, S.; Leostean, C.; Pana, I.O. Green methodology for the preparation of chitosan/graphene nanomaterial through electrochemical exfoliation and its applicability in Sunset Yellow detection. Electrochim. Acta 2018, 283, 578–589. [Google Scholar] [CrossRef]
- Magerusan, L.; Socaci, C.; Coros, M.; Pogacean, F.; Rosu, M.-C.; Gergely, S.; Pruneanu, S.; Leustean, C.; Pana, I.O. Electrochemical Platform based on Nitrogen-doped Graphene / Chitosan Nanocomposite for Selective Pb2+ Detection. Nanotechnology 2017, 28, 114001. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lee, W.J.; Kim, S.O.; Kim, Y.H. Theory, Synthesis, and Oxygen Reduction Catalysis of Fe-Porphyrin-Like Carbon Nanotube. Phys. Rev. Lett. 2011, 106, 175502. [Google Scholar] [CrossRef] [Green Version]
- Brett, C.M.A.; Oliveira-Brett, A.M. Electrochemistry, Principles, Methods and Applications; Oxford University Press: Oxford, UK, 1993; Chapter 9; pp. 174–185. ISBN -10:0198553889. [Google Scholar] [CrossRef]
- Nkosi, D.; Pillay, J.; Ozoemena, K.I.; Nouneh, K.; Oyama, M. Heterogeneous electron transfer kinetics and electrocatalytic behaviour of mixed self-assembled ferrocenes and SWCNT layers. Phys. Chem. Chem. Phys. 2010, 12, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Zoski, C.G. (Ed.) Handbook of Electrochemistry; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 978-0-444-51958-0. [Google Scholar] [CrossRef]
- Su, W.Y.; Wang, S.M.; Cheng, S.H. Electrochemically pretreated screen-printed carbon electrodes for the simultaneous determination of aminophenol isomers. J. Electroanal. Chem. 2011, 651, 166–172. [Google Scholar] [CrossRef]
- Alam, A.U.; Qin, Y.; Catalano, M.; Wang, L.; Kim, M.J.; Howlader, M.M.R.; Hu, N.-X.; Deen, M.J. Tailoring MWCNTs and β-Cyclodextrin for Sensitive Detection of Acetaminophen and Estrogen. ACS Appl. Mater. Interfaces 2018, 10, 21411–21427. [Google Scholar] [CrossRef]
- Hanabaratti, R.M.; Tuwar, S.M.; Nandibewoor, S.T.; Gowda, J.I. Fabrication and characterization of zinc oxide nanoparticles modified glassy carbon electrode for sensitive determination of paracetamol. Chem. Data Collect. 2020, 30, 100540. [Google Scholar] [CrossRef]
- Mahmoud, B.G.; Khairy, M.; Rashwan, F.A.; Banks, C.E. Simultaneous Voltammetric Determination of Acetaminophen and Isoniazid (Hepatotoxicity-Related Drugs) Utilizing Bismuth Oxide Nanorod Modified Screen-Printed Electrochemical Sensing Platforms. Anal. Chem. 2017, 89, 2170–2178. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, H.; Gao, X.; Ma, H. Electrochemical Behavior and Voltammetric Determination of Acetaminophen Based on Glassy Carbon Electrodes Modified with poly(4-Aminobenzoic Acid)/electrochemically Reduced Graphene Oxide Composite Films. Mater. Sci. Eng. C 2014, 45, 21–28. [Google Scholar] [CrossRef]
- Brownson, D.A.C.; Banks, C.E. Interpreting Electrochemistry. In The Handbook of Graphene Electrochemistry; Springer: London, UK, 2014; pp. 23–77. [Google Scholar]
- Guideline ICH. Validation of analytical procedures: Text and methodology. Q2 (R1) 2005, 1, 5. [Google Scholar]
- Liang, W.; Liu, L.; Li, Y.; Ren, H.; Zhu, T.; Xu, Y.; Ye, B.-C. Nitrogen-rich porous carbon modified electrochemical sensor for the detection of acetaminophen. J. Electroanal. Chem. 2019, 855, 113496. [Google Scholar] [CrossRef]
- Tahereh, M.A.; Gholizadeh, M. Simultaneous voltammetric determination of tyrosine and paracetamol using a carbon nanotube-graphene nanosheet nanocomposite modified electrode in human blood serum and pharmaceuticals. Colloids Surf. B Biointerfaces 2013, 103, 84–93. [Google Scholar] [CrossRef]
- Zhang, D.; Qian, J.; Yi, Y.; Kingsford, O.J.; Zhu, G. Nitrogen-doped hollow carbon nanospheres wrapped with MoS2 nanosheets for simultaneous electrochemical determination of acetaminophen and 4-aminophenol. J. Electroanal. Chem. 2019, 847, 113229. [Google Scholar] [CrossRef]
- Zhang, W.; Zong, L.; Liu, S.; Pei, S.; Zhang, Y.; Ding, X.; Jiang, B.; Zhang, Y. An electrochemical sensor based on electro-polymerization of caffeic acid and Zn/Ni-ZIF-8–800 on glassy carbon electrode for the sensitive detection of acetaminophen. Biosens. Bioelectron. 2019, 131, 200–206. [Google Scholar] [CrossRef]
- Kumar, S.P.; Giribabu, K.; Manigandan, R.; Munusamy, S.; Muthamizh, S.; Padmanaban, A.; Dhanasekaran, T.; Suresh, R.; Narayanan, V. Simultaneous determination of paracetamol and 4-aminophenol based on poly(chromium Schiff base complex) modified electrode at nanomolar levels. Electrochim. Acta 2016, 194, 116–126. [Google Scholar] [CrossRef]
- Mehretie, S.; Admassie, S.; Hunde, T.; Tessema, M.; Solomon, T. Simultaneous determination of N-acetyl-p-aminophenol and p-aminophenol with poly(3,4-ethylenedioxythiophene) modified glassy carbon electrode. Talanta 2011, 85, 1376–1382. [Google Scholar] [CrossRef]
- Fanjul-Bolado, P.; Lamas-Ardisana, P.J.; Hernández-Santos, D.; Costa-García, A. Electrochemical study and flow injection analysis of paracetamol in pharmaceutical formulations based on screen-printed electrodes and carbon nanotubes. Anal. Chim. Acta 2009, 638, 133–138. [Google Scholar] [CrossRef]
- Cofan, C.; Radovan, C. Simultaneous chronoamperometric sensing of ascorbic acid and acetaminophen at a boron-doped diamond electrode. Sensors 2008, 8, 3952–3969. [Google Scholar] [CrossRef]
- Liu, R.; Zeng, X.; Liu, J.; Luo, J.; Zheng, Y.; Liu, X. A glassy carbon electrode modified with an amphiphilic, electroactive and photosensitive polymer and with multi-walled carbon nanotubes for simultaneous determination of dopamine and paracetamol. Microchim. Acta 2016, 183, 1543–1551. [Google Scholar] [CrossRef]
- Kang, X.; Wang, J.; Wu, H.; Liu, J.; Aksay, I.A.; Lin, Y. A graphene-based electrochemical sensor for sensitive detection of paracetamol. Talanta 2010, 81, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Xie, K.; Zheng, Y.; Zhang, L.; Su, W. Graphene Ink Film Based Electrochemical Detector for Paracetamol Analysis. Electronics 2018, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Okoth, O.K.; Yan, K.; Liu, L.; Zhang, J. Simultaneous electrochemical determination of paracetamol and diclofenac based on poly(diallyldimethylammonium chloride) functionalized graphene. Electroanalysis 2016, 28, 76–82. [Google Scholar] [CrossRef]
- Zidan, M.; Zawawi, R.M.; Erhayem, M.; Salhin, A. Electrochemical Detection of Paracetamol Using Graphene Oxide-Modified Glassy Carbon Electrode. Int. J. Electrochem. Sci. 2014, 9, 7605–7613. [Google Scholar]
- Movlaee, K.; Beitollahi, H.; Reza Ganjali, M.; Norouzi, P. Electrochemical platform for simultaneous determination of levodopa, acetaminophen and tyrosine using a graphene and ferrocene modified carbon paste electrode. Microchim. Acta 2017, 184, 3281–3289. [Google Scholar] [CrossRef]
- Eisele, A.P.P.; Valezi, C.F.; Sartori, E.R. Exploiting the high oxidation potential of carisoprodol on a boron-doped diamond electrode: An improved method for its simultaneous determination with acetaminophen and caffeine. Analyst 2017, 142, 3514–3521. [Google Scholar] [CrossRef]
- Baccarin, M.; Santos, F.A.; Vicentini, F.C.; Zucolotto, V.; Janegitz, B.C.; Fatibello-Filho, O. Electrochemical sensor based on reduced graphene oxide/carbon black/chitosan composite for the simultaneous determination of dopamine and paracetamol concentrations in urine samples. J. Electroanal. Chem. 2017, 799, 436–443. [Google Scholar] [CrossRef]
- Liu, X.; Shangguan, E.; Li, J.; Ning, S.; Guo, L.; Li, Q. A novel electrochemical sensor based on FeS anchored reduced graphene oxide nanosheets for simultaneous determination of dopamine and acetaminophen. Mater. Sci. Eng. C 2017, 70, 628–636. [Google Scholar] [CrossRef]
- Asadpour-Zeynali, K.; Amini, R. Nanostructured hexacyanoferrate intercalated Ni/Al layered double hydroxide modified electrode as a sensitive electrochemical sensor for paracetamol determination. Electroanalysis 2017, 29, 635–642. [Google Scholar] [CrossRef]
- Liu, L.; Lv, H.; Wang, C.; Ao, Z.; Wang, G. Fabrication of the protonated graphitic carbon nitride nanosheets as enhanced electrochemical sensing platforms for hydrogen peroxide and paracetamol detection. Electrochim. Acta 2016, 206, 259–269. [Google Scholar] [CrossRef]
- Yu, S.; Li, H.; Li, G.; Niu, L.; Liu, W.; Di, X. Reduced graphene oxide-supported gold dendrite for electrochemical sensing of acetaminophen. Talanta 2018, 184, 244–250. [Google Scholar] [CrossRef]
- Meenakshi, S.; Pandian, K.; Gopinath, S.C.B. Quantitative simultaneous determination of pentoxifylline and paracetamol in drug and biological samples at graphene nanoflakes modified electrode. J. Taiwan Inst. Chem. Eng. 2020, 107, 15–23. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2θ | D | d | n | % | |
|---|---|---|---|---|---|
| FLG | 21.76 | 13.28 | 4.08 | 3.25 | 76.43 |
| MLG | 26.45 | 93.06 | 3.37 | 27.61 | 23.57 |
| Modified Electrode | Method | Linear Range (µM) | LOD (µM) | Reference |
|---|---|---|---|---|
| MWCNTs-GNS/GCE MWCNTs-GNS—multiwalled carbon nanotubes-graphene nanosheet nanocomposite GCE—glassy carbon electrode | DPV | 90–112 | 9 | [68] |
| MWCNT-β-CD/GCE MWCNT-β-CD—multiwalled carbon nanotubes-β-cyclodextrin GCE—glassy carbon electrode | DPV | 0.05–1 1–300 | 0.0115 | [30] |
| MoS2@NHCSsx/GCE MoS2@NHCSs—nitrogen-doped hollow carbon spheres wrapped with MoS2 nanosheets GCE—glassy carbon electrode | DPV | 0.05–20 | 0.02 | [69] |
| PCA@Zn/Ni-ZIF-8–800/GCE PCA@Zn/Ni-ZIF-8–800—poly caffeic acid (PCA)-Metal-organic frameworks Zn/Ni-ZIF-8–800 GCE—glassy carbon electrode | DPV | 0.08–1000 | 0.029 | [70] |
| Cr-SBC/GCE Cr-SBC—poly(chromium Schiff base complex) GCE—glassy carbon electrode | DPV | 0.008–0.125 | 0.0056 | [71] |
| PEDOT/GCE PEDOT—poly(3,4-ethylenedioxythiophene) GCE—glassy carbon electrode | DPV | 1–100 | 0.4 | [72] |
| GO-Y/CPE GO-Y CPE—carbon paste electrode | DPV | 7–400 | 1.45 | [32] |
| CNTs/SPCE CNTs—carbon nanotubes SPCE—screen-printed electrode | CV | 2.5–1000 | 0.1 | [73] |
| BDDE BDDE—boron-doped diamond electrode | CV | 10–100 | 0.97 | [74] |
| MWCNT-BPVCM/GCE MWCNT-BPVCM—branched amphiphilic photo-sensitive and electroactive polymer-multiwalled carbon nanotubes hybrid GCE—glassy carbon electrode | CV | 5–1000 | 3.5 | [75] |
| Graphene/GCE GCE—glassy carbon electrode | CV | 0.1–20 | 0.032 | [76] |
| GI/GCE GI—graphene ink GCE—glassy carbon electrode | CV | 10–500 | 2.7 | [77] |
| PDDA-GR/GCE PDDA-GR—poly(diallyl- dimethylammonium chloride) functionalized graphene GCE—glassy carbon electrode | CV | 20–200 | 0.221 | [78] |
| GO/GCE GO—graphene oxide GCE—glassy carbon electrode | SWV | 0.05–1 | 0.049 | [79] |
| EFTAG/CPE EFTAG—ethyl 2-(4-ferrocenyl-[1,2,3]triazol-1-yl)-graphene CPE—carbon paste electrode | SWV | 1–150 | 0.5 | [80] |
| BDDE BDDE—boron-doped diamond electrode | SWV | 2.99–283 | 0.768 | [81] |
| RGO-CB-CTS/GCE RGO-CB-CTS—reduced graphene oxide-carbon black-chitosan GCE—glassy carbon electrode | SWV | 2.8–19 | 0.053 | [82] |
| FeS/rGO/GCE FeS/rGO—FeS anchored reduced graphene oxide nanosheets GCE—glassy carbon electrode | Amperometry | 2–8 | 0.1 | [83] |
| Ni-Al-HCF LDH/GCE Ni-Al-HCF LDH—Ni-Al-hexacyanoferrate (III)-layered double hydroxides GCE—glassy carbon electrode | Amperometry | 3–1500 | 0.8 | [84] |
| Protonated g-C3N4/CTS-GCE Protonated g-C3N4 /CTS graphitic carbon nitride nanosheets GCE—glassy carbon electrode | Amperometry | 1.7–2020 | 0.15 | [85] |
| RGO-gold dendrite/GCE RGO-gold dendrite—reduced graphene oxide-supported gold dendrite GCE—glassy carbon electrode | Amperometry | 0.07–3000 | 0.005 | [86] |
| GrNF/GCE GrNF—graphene nanoflakes GCE—glassy carbon electrode | Amperometry | 0.001–150 | 0.004 | [87] |
| exf-NGr/GCE | Amperometry | 0.009–28.8 | 0.003 | current work |
| exf-NGr/GCE | CV | 0.1–100 | 0.03 | current work |
| Tablet (500 mg AMP/Tablet) Analyzed Solution Concentration 13.2 µM | Detected AMP Content (µM) | * Recovery (%) | |
|---|---|---|---|
| CV | UV-Vis | ||
| Brand 1 | 12.9 | 14.1 | 97.72 |
| Brand 2 | 12.6 | 13.7 | 95.45 |
| Brand 3 | 11.18 | 13.3 | 84.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magerusan, L.; Pogacean, F.; Pruneanu, S. Enhanced Acetaminophen Electrochemical Sensing Based on Nitrogen-Doped Graphene. Int. J. Mol. Sci. 2022, 23, 14866. https://doi.org/10.3390/ijms232314866
Magerusan L, Pogacean F, Pruneanu S. Enhanced Acetaminophen Electrochemical Sensing Based on Nitrogen-Doped Graphene. International Journal of Molecular Sciences. 2022; 23(23):14866. https://doi.org/10.3390/ijms232314866
Chicago/Turabian StyleMagerusan, Lidia, Florina Pogacean, and Stela Pruneanu. 2022. "Enhanced Acetaminophen Electrochemical Sensing Based on Nitrogen-Doped Graphene" International Journal of Molecular Sciences 23, no. 23: 14866. https://doi.org/10.3390/ijms232314866