



The STIM1/2-Regulated Calcium Homeostasis Is Impaired in Hippocampal Neurons of the 5xFAD Mouse Model of Alzheimer’s Disease
 , , and
, , and
Abstract
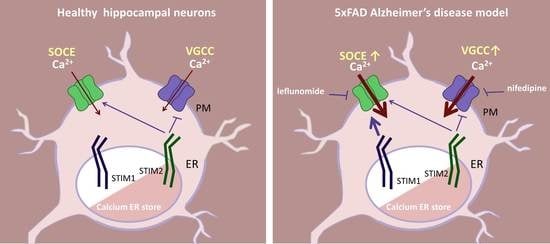
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
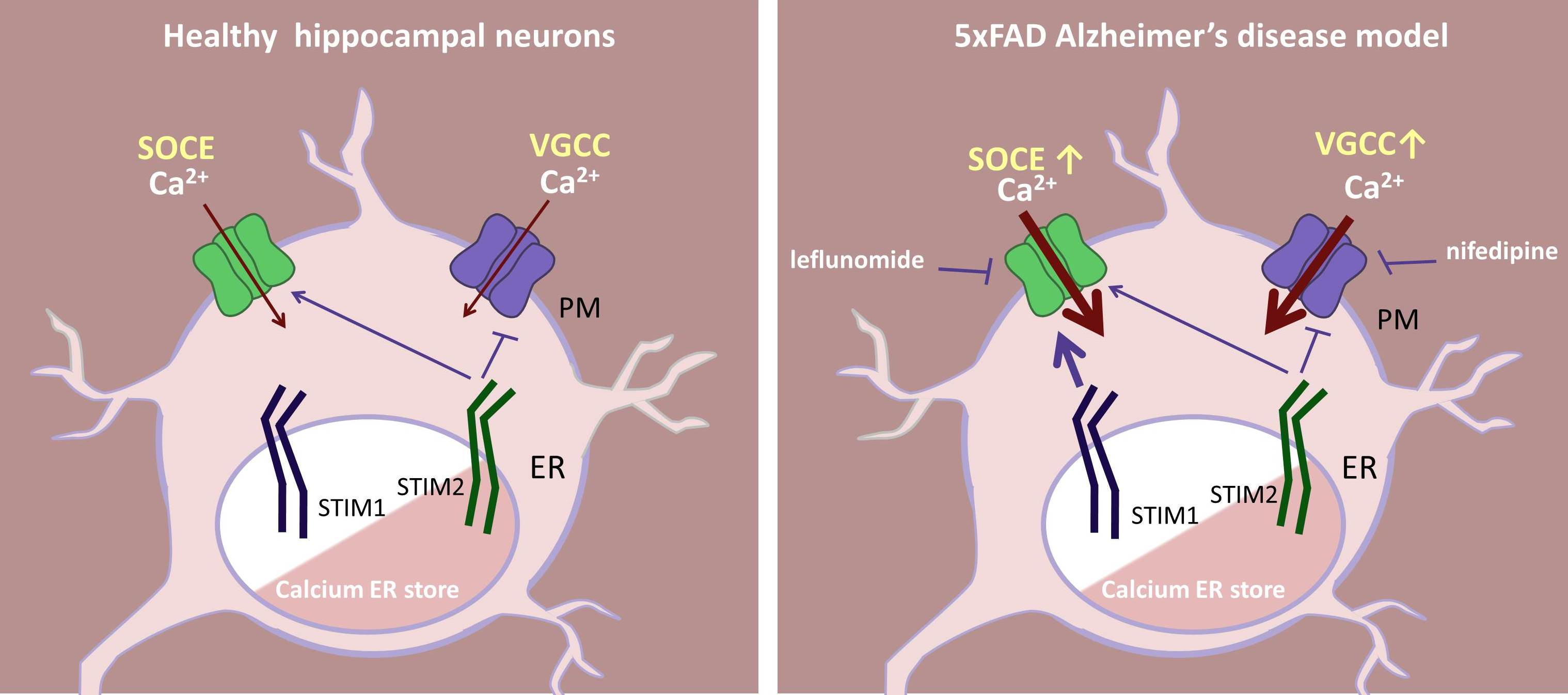
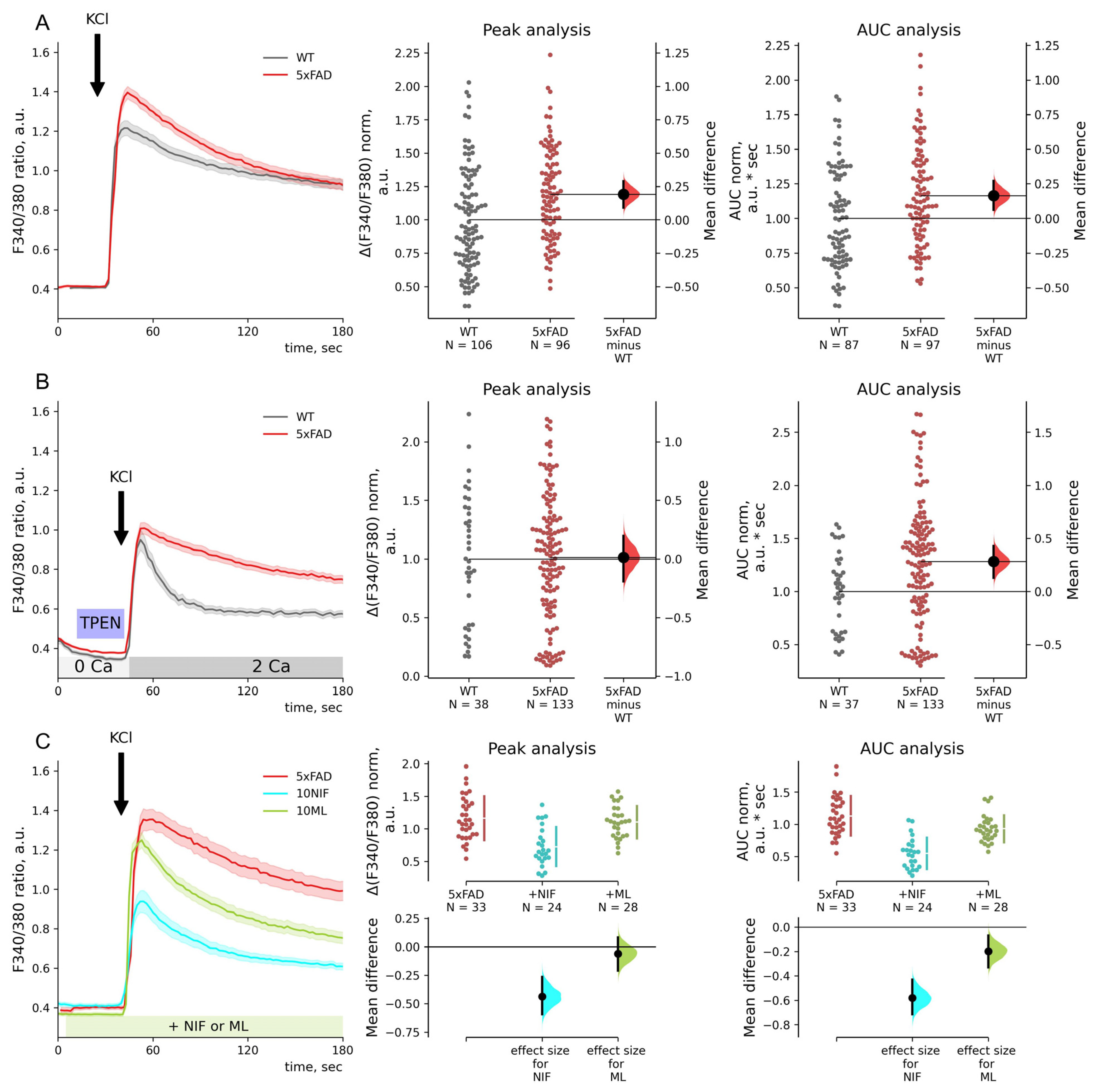
2.1. Store-Operated Calcium Entry Is Enhanced in 5xFAD Hippocampal Neurons
2.2. FDA-Approved Drugs Reduce SOC Entry in 5xFAD Mouse Neurons
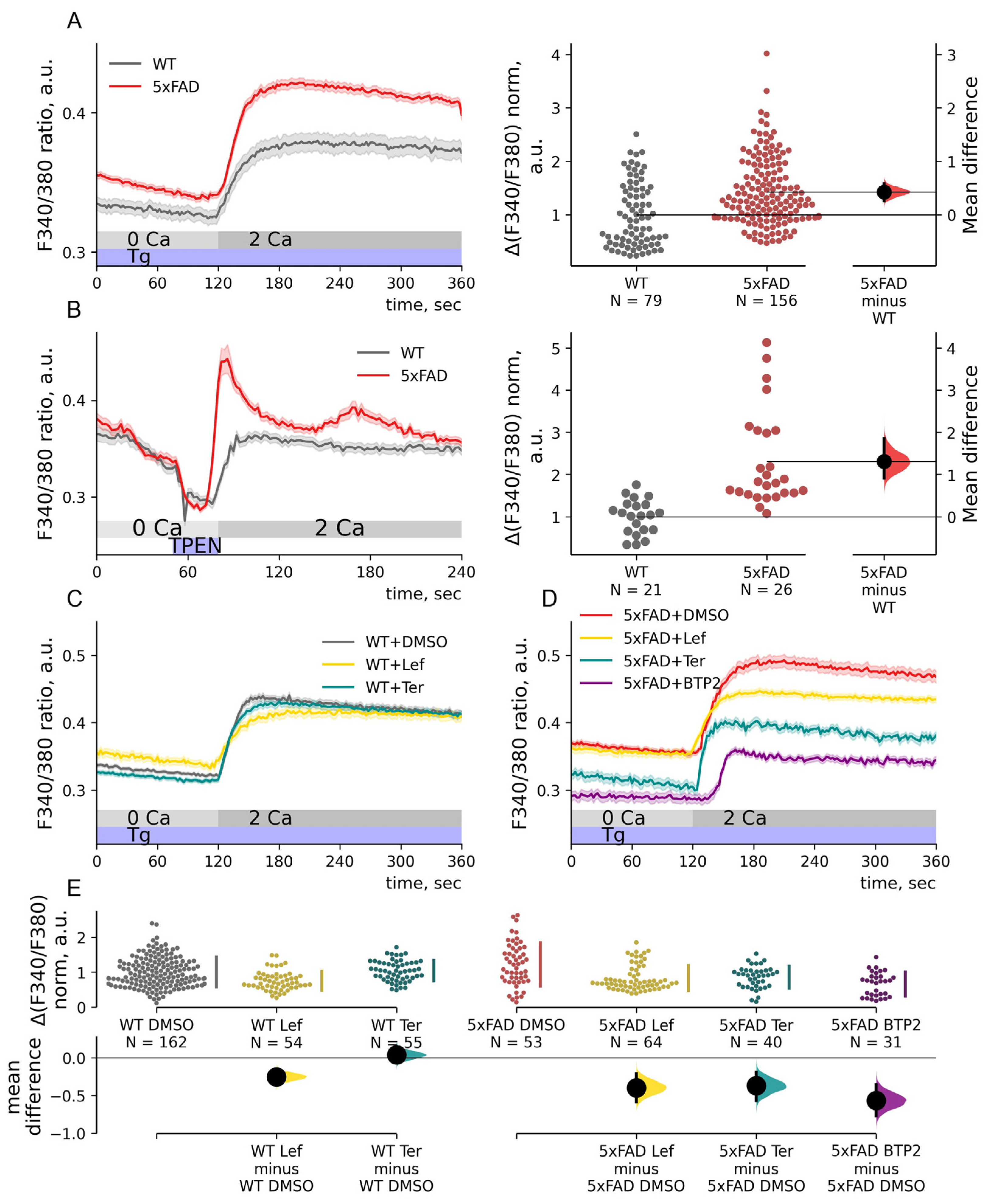
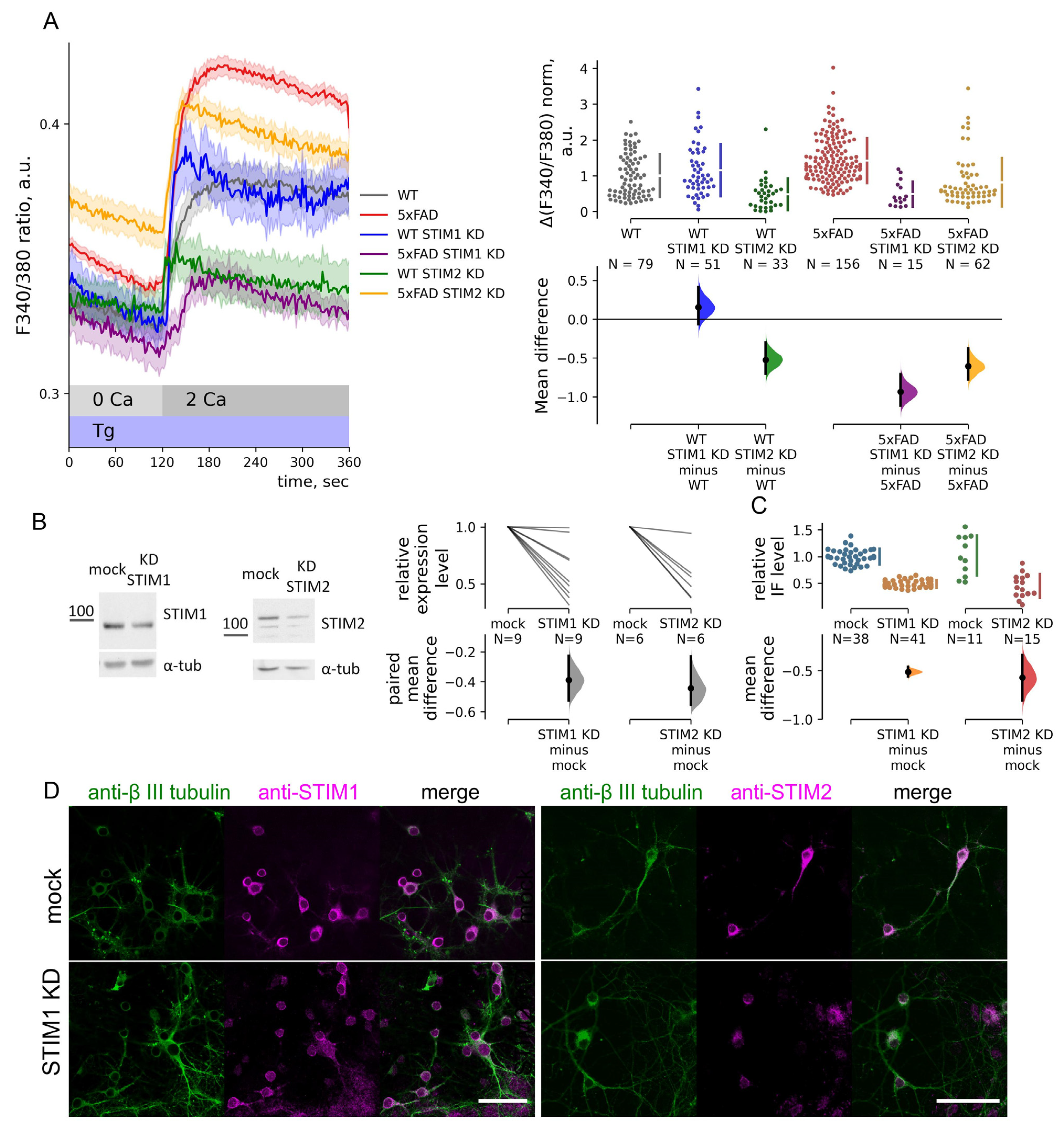
2.3. SOC Entry Enhancement in 5xFAD Mouse Neurons Is Associated with Changes in the Passive Leak but Not with Expression Levels of STIMs
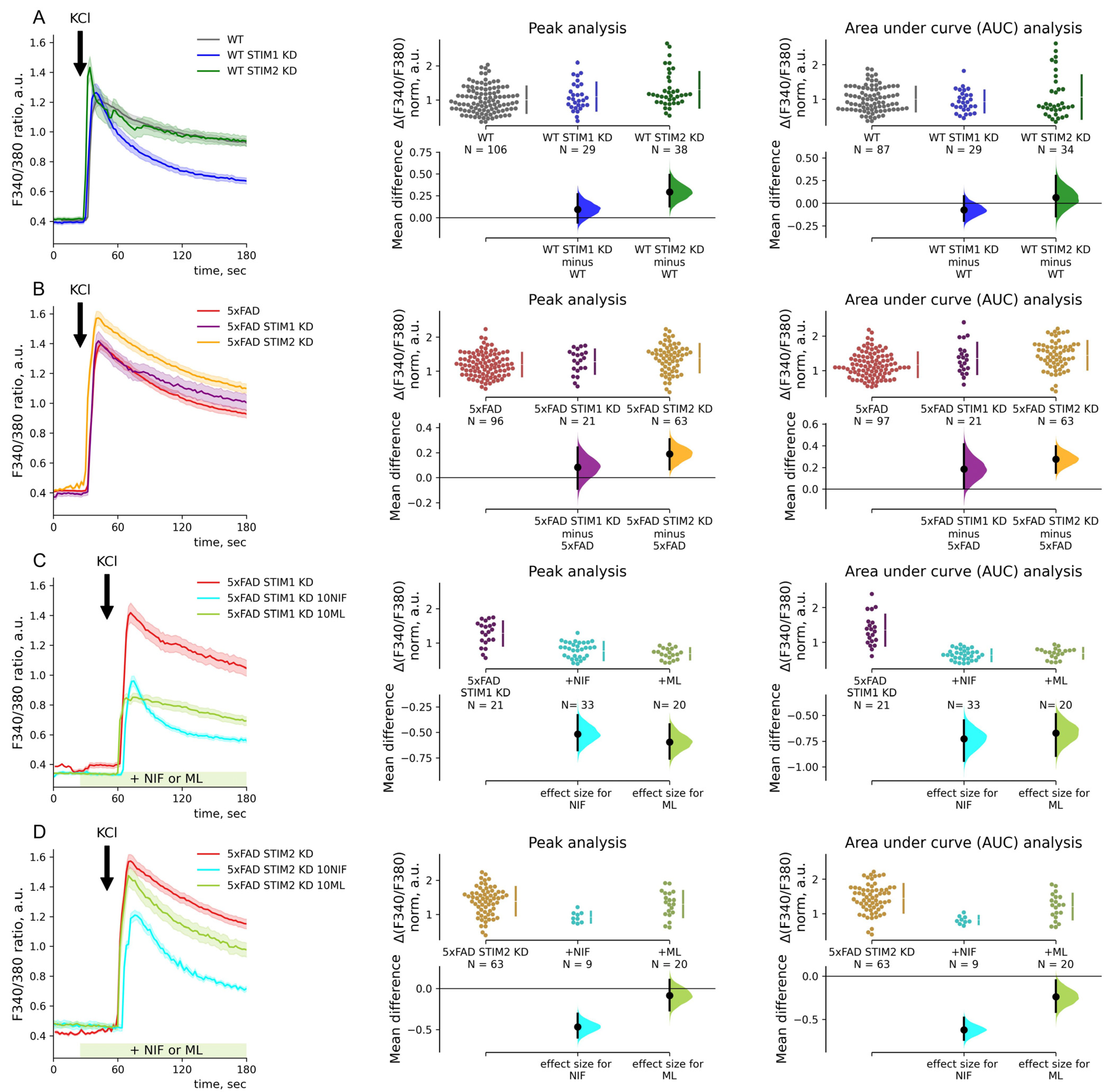
2.4. STIM1 Contribution to SOC Entry Is Increased in 5xFAD Mouse Neurons
2.5. Depolarization-Induced Calcium Entry Is Enhanced in 5xFAD Neurons
3. Discussion
3.1. STIM1 Drives Increased SOC Entry in Hippocampal Neurons of the 5xFAD Mouse Model
3.2. STIM2 Regulates VGC Entry in 5xFAD Neurons with Replete ER Calcium Store
4. Materials and Methods
4.1. Animals
4.2. Cell Cultures
4.3. Lentiviral Infection
4.4. Western Blotting
4.5. Immunostaining
4.6. Calcium Imaging
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Eldik, L.V.; et al. Intraneuronal Beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [Green Version]
- Ghoweri, A.O.; Ouillette, L.; Frazier, H.N.; Anderson, K.L.; Lin, R.-L.; Gant, J.C.; Parent, R.; Moore, S.; Murphy, G.G.; Thibault, O. Electrophysiological and Imaging Calcium Biomarkers of Aging in Male and Female 5×FAD Mice. J. Alzheimers Dis. 2020, 78, 1419–1438. [Google Scholar] [CrossRef]
- Reitz, C. Alzheimer’s Disease and the Amyloid Cascade Hypothesis: A Critical Review. J. Alzheimers Dis. 2012, 2012, 369808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, P.-P.; Cao, L.-L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci 2021, 22, 5900. [Google Scholar] [CrossRef]
- Shim, H.S.; Horner, J.W.; Wu, C.-J.; Li, J.; Lan, Z.D.; Jiang, S.; Xu, X.; Hsu, W.-H.; Zal, T.; Flores, I.I.; et al. Telomerase Reverse Transcriptase Preserves Neuron Survival and Cognition in Alzheimer’s Disease Models. Nat. Aging 2021, 1, 1162–1174. [Google Scholar] [CrossRef]
- Pilat, D.; Paumier, J.-M.; García-González, L.; Louis, L.; Stephan, D.; Manrique, C.; Khrestchatisky, M.; Di Pasquale, E.; Baranger, K.; Rivera, S. MT5-MMP Promotes Neuroinflammation, Neuronal Excitability and Aβ Production in Primary Neuron/Astrocyte Cultures from the 5xFAD Mouse Model of Alzheimer’s Disease. J. Neuroinflammation 2022, 19, 65. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 Signaling in Cortical Neurons of Knock-in Mice Expressing an Alzheimer’s-Linked Mutation in Presenilin1 Results in Exaggerated Ca2+ Signals and Altered Membrane Excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.F.; Hitt, B.; Green, K.N.; Oddo, S.; LaFerla, F.M. Enhanced Caffeine-Induced Ca2+ Release in the 3xTg-AD Mouse Model of Alzheimer’s Disease. J. Neurochem. 2005, 94, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Lerdkrai, C.; Asavapanumas, N.; Brawek, B.; Kovalchuk, Y.; Mojtahedi, N.; Moral, M.O.D.; Garaschuk, O. Intracellular Ca2+ Stores Control in Vivo Neuronal Hyperactivity in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1279–E1288. [Google Scholar] [CrossRef] [PubMed]
- Rigotto, G.; Zentilin, L.; Pozzan, T.; Basso, E. Effects of Mild Excitotoxic Stimulus on Mitochondria Ca2+ Handling in Hippocampal Cultures of a Mouse Model of Alzheimer’s Disease. Cells 2021, 10, 2046. [Google Scholar] [CrossRef]
- Popugaeva, E.; Bezprozvanny, I.; Chernyuk, D. Reversal of Calcium Dysregulation as Potential Approach for Treating Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. Int. J. Mol. Sci. 2020, 21, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Day, D.H. Calmodulin Binding Proteins and Alzheimer’s Disease: Biomarkers, Regulatory Enzymes and Receptors That Are Regulated by Calmodulin. Int. J. Mol. Sci. 2020, 21, E7344. [Google Scholar] [CrossRef]
- Grekhnev, D.A.; Kaznacheyeva, E.V.; Vigont, V.A. Patient-Specific IPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling. Int. J. Mol. Sci. 2022, 23, 624. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The Role of Mitochondrial Calcium Homeostasis in Alzheimer’s and Related Diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef]
- Kumar, A. Calcium Signaling During Brain Aging and Its Influence on the Hippocampal Synaptic Plasticity. Adv. Exp. Med. Biol. 2020, 1131, 985–1012. [Google Scholar] [CrossRef]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium Responses in Fibroblasts from Asymptomatic Members of Alzheimer’s Disease Families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [Green Version]
- LaFerla, F.M. Calcium Dyshomeostasis and Intracellular Signalling in Alzheimer’s Disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, W. Ca 2+ Homeostasis Dysregulation in Alzheimer’s Disease: A Focus on Plasma Membrane and Cell Organelles. FASEB J. 2019, 33, 6697–6712. [Google Scholar] [CrossRef] [PubMed]
- Serwach, K.; Gruszczynska-Biegala, J. Target Molecules of STIM Proteins in the Central Nervous System. Front. Mol. Neurobiol. 2020, 13, 617422. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. Store-Operated Calcium Channels: From Function to Structure and Back Again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalygin, A.; Kolesnikov, D.; Glushankova, L.; Gusev, K.; Skopin, A.; Skobeleva, K.; Kaznacheyeva, E.V. Role of STIM2 and Orai Proteins in Regulating TRPC1 Channel Activity upon Calcium Store Depletion. Cell Calcium 2021, 97, 102432. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.-J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sun, S.; Herreman, A.A.; Strooper, B.D.; Bezprozvanny, I. Role of Presenilins in Neuronal Calcium Homeostasis. J. Neurosci. 2010, 30, 8566–8580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sun, S.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Tacer, K.F.; Bezprozvanny, I. Store-Operated Calcium Channel Complex in Postsynaptic Spines: A New Therapeutic Target for Alzheimer’s Disease Treatment. J. Neurosci. 2016, 36, 11837–11850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; Hernando-Pérez, E.; López-Vázquez, S.; Núñez, J.; Villalobos, C.; Núñez, L. Remodeling of Intracellular Ca2+ Homeostasis in Rat Hippocampal Neurons Aged In Vitro. Int. J. Mol. Sci. 2020, 21, 1549. [Google Scholar] [CrossRef] [PubMed]
- Ryazantseva, M.; Goncharova, A.; Skobeleva, K.; Erokhin, M.; Methner, A.; Georgiev, P.; Kaznacheyeva, E. Presenilin-1 Delta E9 Mutant Induces STIM1-Driven Store-Operated Calcium Channel Hyperactivation in Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 4667–4680. [Google Scholar] [CrossRef]
- Ryazantseva, M.; Skobeleva, K.; Kaznacheyeva, E. Familial Alzheimer’s Disease-Linked Presenilin-1 Mutation M146V Affects Store-Operated Calcium Entry: Does Gain Look like Loss? Biochimie 2013, 95, 1506–1509. [Google Scholar] [CrossRef]
- Chernyuk, D.; Zernov, N.; Kabirova, M.; Bezprozvanny, I.; Popugaeva, E. Antagonist of Neuronal Store-Operated Calcium Entry Exerts Beneficial Effects in Neurons Expressing PSEN1ΔE9 Mutant Linked to Familial Alzheimer Disease. Neuroscience 2019, 410, 118–127. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 Is a Feedback Regulator That Stabilizes Basal Cytosolic and Endoplasmic Reticulum Ca2+ Levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruszczynska-Biegala, J.; Pomorski, P.; Wisniewska, M.B.; Kuznicki, J. Differential Roles for STIM1 and STIM2 in Store-Operated Calcium Entry in Rat Neurons. PLoS ONE 2011, 6, e19285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC Channel Activator STIM1 Binds and Inhibits L-Type Voltage-Gated Calcium Channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The Calcium Store Sensor, STIM1, Reciprocally Controls Orai and CaV1.2 Channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.; Biet, M.; Simard, É.; Béliveau, É.; Francoeur, N.; Guillemette, G.; Dumaine, R.; Grandbois, M.; Boulay, G. STIM1 Participates in the Contractile Rhythmicity of HL-1 Cells by Moderating T-Type Ca2+ Channel Activity. Biochim. Biophys. Acta -Mol. Cell Res. 2013, 1833, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Majewski, L.; Kuznicki, J. SOCE in Neurons: Signaling or Just Refilling? Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 1940–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 Deficiency Is Linked to Alzheimer’s Disease and Triggers Cell Death in SH-SY5Y Cells by Upregulation of L-Type Voltage-Operated Ca2+ Entry. J. Mol. Med. 2018, 96, 1061–1079. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Pchitskaya, E.; Speshilova, A.; Alexandrov, S.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. STIM2 Protects Hippocampal Mushroom Spines from Amyloid Synaptotoxicity. Mol. Neurodegener. 2015, 10, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, A.M.; Fasolato, C.; Pozzan, T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: A study using simultaneous measurements of ICRAC and intraluminal [Ca2+]. J. Cell Biol. 1998, 140, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Rahman, T. Unveiling Some FDA-Approved Drugs as Inhibitors of the Store-Operated Ca2+ Entry Pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breedveld, F.C.; Dayer, J.-M. Leflunomide: Mode of Action in the Treatment of Rheumatoid Arthritis. Ann. Rheum Dis. 2000, 59, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Skibinska-Kijek, A.; Wisniewska, M.B.; Gruszczynska-Biegala, J.; Methner, A.; Kuznicki, J. Immunolocalization of STIM1 in the Mouse Brain. Acta Neurobiol. Exp. 2009, 69, 413–428. [Google Scholar]
- Chen-Engerer, H.J.; Hartmann, J.; Karl, R.M.; Yang, J.; Feske, S.; Konnerth, A. Two Types of Functionally Distinct Ca2+ Stores in Hippocampal Neurons. Nat. Commun. 2019, 10, 3223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualdani, R.; Gailly, P. How TRPC Channels Modulate Hippocampal Function. Int. J. Mol. Sci. 2020, 21, 3915. [Google Scholar] [CrossRef]
- Secondo, A.; Bagetta, G.; Amantea, D. On the Role of Store-Operated Calcium Entry in Acute and Chronic Neurodegenerative Diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarski, L.; Pomorski, P.; Szybinska, A.; Drab, M.; Skibinska-Kijek, A.; Gruszczynska-Biegala, J.; Kuznicki, J. Presenilin-Dependent Expression of STIM Proteins and Dysregulation of Capacitative Ca2+ Entry in Familial Alzheimer’s Disease. Biochim. Biophys. Acta 2009, 1793, 1050–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.-F.; Hao, Y.-H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin Mutations Linked to Familial Alzheimer’s Disease Reduce Endoplasmic Reticulum and Golgi Apparatus Calcium Levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Lee, C.S.-K.; Cheng, W.-H.; Lai, K.-O.; Foskett, J.K.; Cheung, K.-H. Familial Alzheimer’s Disease–Associated Presenilin 1 Mutants Promote γ-Secretase Cleavage of STIM1 to Impair Store-Operated Ca2+ Entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alić, I.; Kosi, N.; Kapuralin, K.; Gorup, D.; Gajović, S.; Pochet, R.; Mitrečić, D. Neural Stem Cells from Mouse Strain Thy1 YFP-16 Are a Valuable Tool to Monitor and Evaluate Neuronal Differentiation and Morphology. Neurosci. Lett. 2016, 634, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ Dysregulation in Neurons from Transgenic Mice Expressing Mutant Presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer Disease–Linked Mutations Specifically Disrupt Ca2+ Leak Function of Presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.-H.; Mei, L.; Mak, D.-O.D.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-Function Enhancement of IP3 Receptor Modal Gating by Familial Alzheimer’s Disease-Linked Presenilin Mutants in Human Cells and Mouse Neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s Disease-Linked Presenilin Mutants and Intracellular Ca2+ Handling: A Single-Organelle, FRET-Based Analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjö, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative Splicing Converts STIM2 from an Activator to an Inhibitor of Store-Operated Calcium Channels. J. Cell Biol. 2015, 209, 653–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, G.; Jarzembowski, L.; Schwarz, Y.; Poth, V.; Konrad, M.; Knapp, M.L.; Schwär, G.; Lauer, A.A.; Grimm, M.O.W.; Alansary, D.; et al. A Short Isoform of STIM1 Confers Frequency-Dependent Synaptic Enhancement. Cell Rep. 2021, 34, 108844. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-Y.; He, Z.-Y.; Yang, L.-J.; Zhang, M.; Xing, D.; Xiao, Z.-C. The Ca2+ Channel Inhibitor 2-APB Reverses β-Amyloid-Induced LTP Deficit in Hippocampus by Blocking BAX and Caspase-3 Hyperactivation. Br. J. Pharmacol. 2015, 172, 2273–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coon, A.L.; Wallace, D.R.; Mactutus, C.F.; Booze, R.M. L-Type Calcium Channels in the Hippocampus and Cerebellum of Alzheimer’s Disease Brain Tissue. Neurobiol. Aging 1999, 20, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Thibault, O.; Pancani, T.; Landfield, P.W.; Norris, C.M. Reduction in Neuronal L-Type Calcium Channel Activity in a Double Knock-in Mouse Model of Alzheimer’s Disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 546–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Mattson, M.P. L-Type Ca2+ Currents at CA1 Synapses, but Not CA3 or Dentate Granule Neuron Synapses, Are Increased in 3xTgAD Mice in an Age-Dependent Manner. Neurobiol. Aging 2014, 35, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, M.; Hiller, A.J.; Pham, L.; McGuire, M.J.; Iadecola, C.; Wang, G. Amyloid-Beta Modulates Low-Threshold Activated Voltage-Gated L-Type Calcium Channels of Arcuate Neuropeptide Y Neurons Leading to Calcium Dysregulation and Hypothalamic Dysfunction. J. Neurosci. 2019, 39, 8816–8825. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiao, J.-J.; Su, Q.; Hölscher, C.; Zhang, J.; Yan, X.-D.; Zhao, H.-M.; Cai, H.-Y.; Qi, J.-S. A GLP-1/GIP/Gcg Receptor Triagonist Improves Memory Behavior, as Well as Synaptic Transmission, Neuronal Excitability and Ca2+ Homeostasis in 3xTg-AD Mice. Neuropharmacology 2020, 170, 108042. [Google Scholar] [CrossRef]
- Harraz, O.F.; Altier, C. STIM1-Mediated Bidirectional Regulation of Ca(2+) Entry through Voltage-Gated Calcium Channels (VGCC) and Calcium-Release Activated Channels (CRAC). Front. Cell Neurosci. 2014, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wang, J.; Peng, G.; Shimoda, L.A.; Sylvester, J.T. Knockdown of Stromal Interaction Molecule 1 Attenuates Store-Operated Ca2+ Entry and Ca2+ Responses to Acute Hypoxia in Pulmonary Arterial Smooth Muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L17–L25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigont, V.A.; Grekhnev, D.A.; Lebedeva, O.S.; Gusev, K.O.; Volovikov, E.A.; Skopin, A.Y.; Bogomazova, A.N.; Shuvalova, L.D.; Zubkova, O.A.; Khomyakova, E.A.; et al. STIM2 Mediates Excessive Store-Operated Calcium Entry in Patient-Specific IPSC-Derived Neurons Modeling a Juvenile Form of Huntington’s Disease. Front. Cell Dev. Biol. 2021, 9, 625231. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 Regulates Capacitive Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A Conserved, Lipid-Mediated Sorting Mechanism of Yeast Ist2 and Mammalian STIM Proteins to the Peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shih, H.-P.; Vigont, V.; Hrdlicka, L.; Diggins, L.; Singh, C.; Mahoney, M.; Chesworth, R.; Shapiro, G.; Zimina, O.; et al. Neuronal Store-Operated Calcium Entry Pathway as a Novel Therapeutic Target for Huntington’s Disease Treatment. Chem. Biol. 2011, 18, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelstein, A.D.; Tsuchida, M.A.; Amodaj, N.; Pinkard, H.; Vale, R.D.; Stuurman, N. Advanced Methods of Microscope Control Using ΜManager Software. J. Biol. Methods 2014, 1, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.; Tumkaya, T.; Aryal, S.; Choi, H.; Claridge-Chang, A. Moving beyond P Values: Data Analysis with Estimation Graphics. Nat. Methods 2019, 16, 565–566. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, D.K. What is the proper way to apply the multiple comparison test? Korean J. Anesthesiol. 2018, 71, 353–360. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skobeleva, K.; Shalygin, A.; Mikhaylova, E.; Guzhova, I.; Ryazantseva, M.; Kaznacheyeva, E. The STIM1/2-Regulated Calcium Homeostasis Is Impaired in Hippocampal Neurons of the 5xFAD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 14810. https://doi.org/10.3390/ijms232314810
Skobeleva K, Shalygin A, Mikhaylova E, Guzhova I, Ryazantseva M, Kaznacheyeva E. The STIM1/2-Regulated Calcium Homeostasis Is Impaired in Hippocampal Neurons of the 5xFAD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(23):14810. https://doi.org/10.3390/ijms232314810
Chicago/Turabian StyleSkobeleva, Ksenia, Alexey Shalygin, Elena Mikhaylova, Irina Guzhova, Maria Ryazantseva, and Elena Kaznacheyeva. 2022. "The STIM1/2-Regulated Calcium Homeostasis Is Impaired in Hippocampal Neurons of the 5xFAD Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 23: 14810. https://doi.org/10.3390/ijms232314810