
KARAJ: An Efficient Adaptive Multi-Processor Tool to Streamline Genomic and Transcriptomic Sequence Data Acquisition

 ,
, {kind=link}
Abstract
:1. Introduction
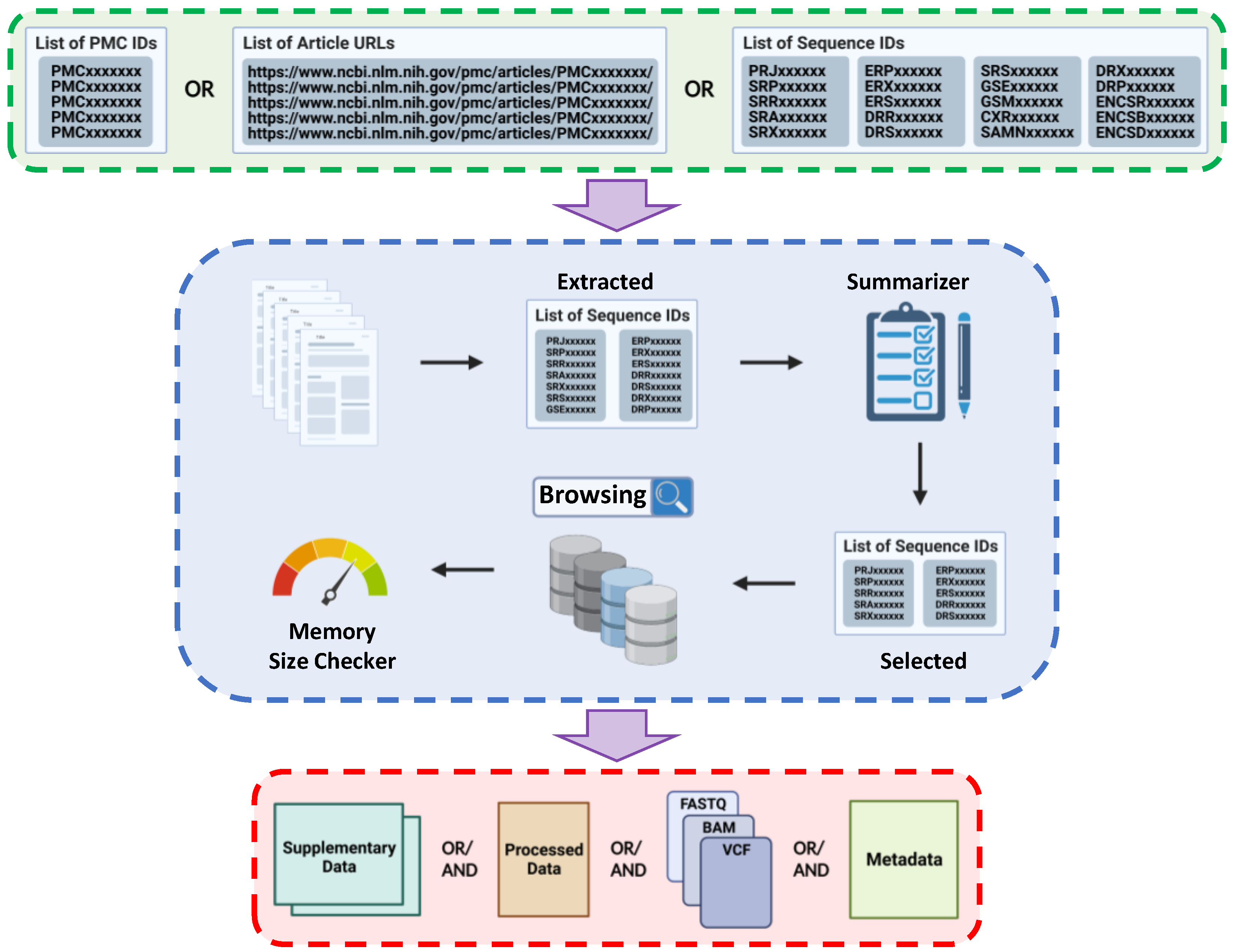
2. Description
3. Installation
4. Tutorial
5. Usage Examples
5.1. Scenario 1
5.2. Scenario 2
5.3. Scenario 3
5.4. Scenario 4
5.5. Scenario 5
5.6. Scenario 6
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stephens, Z.D.; Lee, S.Y.; Faghri, F.; Campbell, R.H.; Zhai, C.; Efron, M.J.; Iyer, R.; Schatz, M.C.; Sinha, S.; Robinson, G.E. Big Data: Astronomical or Genomical? PLoS Biol. 2015, 13, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Afrasiabi, A.; Keane, J.T.; Heng, J.I.; Palmer, E.E.; Lovell, N.H.; Alinejad-Rokny, H. Quantitative neurogenetics: Applications in understanding disease. Biochem. Soc. Trans. 2021, 49, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.C.P.; Mohsen, H.; Yan, C.; Li, S.; Gu, M.; Meyerson, W.; Gerstein, M. Genomics and data science: An application within an umbrella. Genome Biol. 2019, 20, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [Green Version]
- Lipman, D.J.; Pearson, W.R. Rapid and sensitive protein similarity searches. Science 1985, 227, 1435–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M.; on behalf of the International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Cummins, C.; Ahamed, A.; Aslam, R.; Burgin, J.; Devraj, R.; Edbali, O.; Gupta, D.; Harrison, P.W.; Haseeb, M.; Holt, S.; et al. The European Nucleotide Archive in 2021. Nucleic Acids Res. 2022, 50, D106–D110. [Google Scholar] [CrossRef] [PubMed]
- Okido, T.; Kodama, Y.; Mashima, J.; Kosuge, T.; Fujisawa, T.; Ogasawara, O. DNA Data Bank of Japan (DDBJ) update report 2021. Nucleic Acids Res. 2022, 50, D102–D105. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gálvez-Merchán, Á.; Min, K.H.J.; Pachter, L.; Booeshaghi, A.S. Metadata retrieval from sequence databases with ffq. BioRxiv 2022. [Google Scholar] [CrossRef]
- Choudhary, S. pysradb: A Python package to query next-generation sequencing metadata and data from NCBI Sequence Read Archive. F1000Research 2019, 8, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewels, P. SRA-Explorer. Available online: https://github.com/ewels/sra-explorer (accessed on 31 July 2022).
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]
- Cornish, T.C.; Kricka, L.J.; Park, J.Y. A Biopython-based method for comprehensively searching for eponyms in Pubmed. MethodsX 2021, 8, 101264. [Google Scholar] [CrossRef]
- Kans, J. Entrez direct: E-utilities on the UNIX command line. In Entrez Programming Utilities Help [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2022. [Google Scholar]
- Zhu, Y.; Davis, S.; Stephens, R.; Meltzer, P.S.; Chen, Y. GEOmetadb: Powerful alternative search engine for the Gene Expression Omnibus. Bioinformatics 2008, 24, 2798–2800. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Stephens, R.M.; Meltzer, P.S.; Davis, S.R. SRAdb: Query and use public next-generation sequencing data from within R. BMC Bioinform. 2013, 14, 19. [Google Scholar] [CrossRef] [Green Version]
- Sozanska, A.M.; Fletcher, C.; Bihary, D.; Samarajiwa, S.A. SpiderSeqR: An R package for crawling the web of high-throughput multi-omic data repositories for data-sets and annotatio. BioRxiv 2020. [Google Scholar] [CrossRef]
- IBM. What is IBM Aspera Connect? Available online: https://www.ibm.com/docs/en/aspera-on-cloud?topic=client-what-is-aspera-connect (accessed on 31 July 2022).
- Afrasiabi, A.; Parnell, G.P.; Fewings, N.; Schibeci, S.D.; Basuki, M.A.; Chandramohan, R.; Zhou, Y.; Taylor, B.; Brown, D.A.; Swaminathan, S.; et al. Evidence from genome wide association studies implicates reduced control of Epstein-Barr virus infection in multiple sclerosis susceptibility. Genome Med. 2019, 11, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montulli, L.; Blythe, G.; Lavender, C.; Grobe, M.; Rezac, C. Lynx. Available online: https://linux.die.net/man/1/lynx (accessed on 31 July 2022).
- Luceno, I.; Quartulli, A. AXEL—Lightweight CLI Download Accelerator. Available online: https://github.com/axel-download-accelerator/axel (accessed on 31 July 2022).
- Niksic, H.; Cowan, M. wget(1)—Linux Man Page. Available online: https://linux.die.net/man/1/wget (accessed on 31 July 2022).
- Jadhav, B.; Monajemi, R.; Gagalova, K.K.; Ho, D.; Draisma, H.H.M.; van de Wiel, M.A.; Franke, L.; Heijmans, B.T.; van Meurs, J.; Jansen, R.; et al. RNA-Seq in 296 phased trios provides a high-resolution map of genomic imprinting. BMC Biol. 2019, 17, 50. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Liao, J.; Chen, Y.; Zou, C.; Zhang, H.; Cheng, J.; Liu, D.; Li, T.; Zhang, Q.; Li, J.; et al. Single-Cell Transcriptomic Map of the Human and Mouse Bladders. J. Am. Soc. Nephrol. 2019, 30, 2159–2176. [Google Scholar] [CrossRef]
- Lappalainen, T.; Sammeth, M.; Friedlander, M.R.; t Hoen, P.A.; Monlong, J.; Rivas, M.A.; Gonzalez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, L.T.C.; Parnell, G.P.; Afrasiabi, A.; Stewart, G.J.; Swaminathan, S.; Booth, D.R. Transcribed B lymphocyte genes and multiple sclerosis risk genes are underrepresented in Epstein-Barr Virus hypomethylated regions. Genes Immun. 2020, 21, 91–99. [Google Scholar] [CrossRef] [Green Version]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Afrasiabi, A.; Fewings, N.L.; Schibeci, S.D.; Keane, J.T.; Booth, D.R.; Parnell, G.P.; Swaminathan, S. The Interaction of Human and Epstein-Barr Virus miRNAs with Multiple Sclerosis Risk Loci. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Keane, J.T.; Afrasiabi, A.; Schibeci, S.D.; Fewings, N.; Parnell, G.P.; Swaminathan, S.; Booth, D.R. Gender and the Sex Hormone Estradiol Affect Multiple Sclerosis Risk Gene Expression in Epstein-Barr Virus-Infected B Cells. Front. Immunol. 2021, 12, 732694. [Google Scholar] [CrossRef]
- Nasab, R.Z.; Ghamsari, M.R.; Argha, A.; Macphillamy, C.; Beheshti, A.; Alizadehsani, R.; Lovell, N.H.; Alinejad-Rokny, H. Deep Learning in Spatially Resolved Transcriptomics: A Comprehensive Technical View. arXiv 2022, arXiv:2210.04453. [Google Scholar]
- Afrasiabi, A.; Parnell, G.P.; Swaminathan, S.; Stewart, G.J.; Booth, D.R. The interaction of Multiple Sclerosis risk loci with Epstein-Barr virus phenotypes implicates the virus in pathogenesis. Sci. Rep. 2020, 10, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afrasiabi, A.; Alinejad-Rokny, H.; Khosh, A.; Rahnama, M.; Lovell, N.; Xu, Z.; Ebrahimi, D. The low abundance of CpG in the SARS-CoV-2 genome is not an evolutionarily signature of ZAP. Sci. Rep. 2022, 12, 2420. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Shojaei, M.; Wang, Y.; Nalos, M.; McLean, A.; Afrasiabi, A.; Kwan, T.N.; Kuan, W.S.; Zerbib, Y.; Herwanto, V.; et al. Prospective validation study of prognostic biomarkers to predict adverse outcomes in patients with COVID-19: A study protocol. BMJ Open 2021, 11, e044497. [Google Scholar] [CrossRef]
- Keane, J.T.; Afrasiabi, A.; Schibeci, S.D.; Swaminathan, S.; Parnell, G.P.; Booth, D.R. The interaction of Epstein-Barr virus encoded transcription factor EBNA2 with multiple sclerosis risk loci is dependent on the risk genotype. EBioMedicine 2021, 71, 103572. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labani, M.; Beheshti, A.; Lovell, N.H.; Alinejad-Rokny, H.; Afrasiabi, A. KARAJ: An Efficient Adaptive Multi-Processor Tool to Streamline Genomic and Transcriptomic Sequence Data Acquisition. Int. J. Mol. Sci. 2022, 23, 14418. https://doi.org/10.3390/ijms232214418
Labani M, Beheshti A, Lovell NH, Alinejad-Rokny H, Afrasiabi A. KARAJ: An Efficient Adaptive Multi-Processor Tool to Streamline Genomic and Transcriptomic Sequence Data Acquisition. International Journal of Molecular Sciences. 2022; 23(22):14418. https://doi.org/10.3390/ijms232214418
Chicago/Turabian StyleLabani, Mahdieh, Amin Beheshti, Nigel H. Lovell, Hamid Alinejad-Rokny, and Ali Afrasiabi. 2022. "KARAJ: An Efficient Adaptive Multi-Processor Tool to Streamline Genomic and Transcriptomic Sequence Data Acquisition" International Journal of Molecular Sciences 23, no. 22: 14418. https://doi.org/10.3390/ijms232214418