
Variant Enrichment Analysis to Explore Pathways Disruption in a Necropsy Series of Asbestos-Exposed Shipyard Workers
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Study and Populations Characteristics
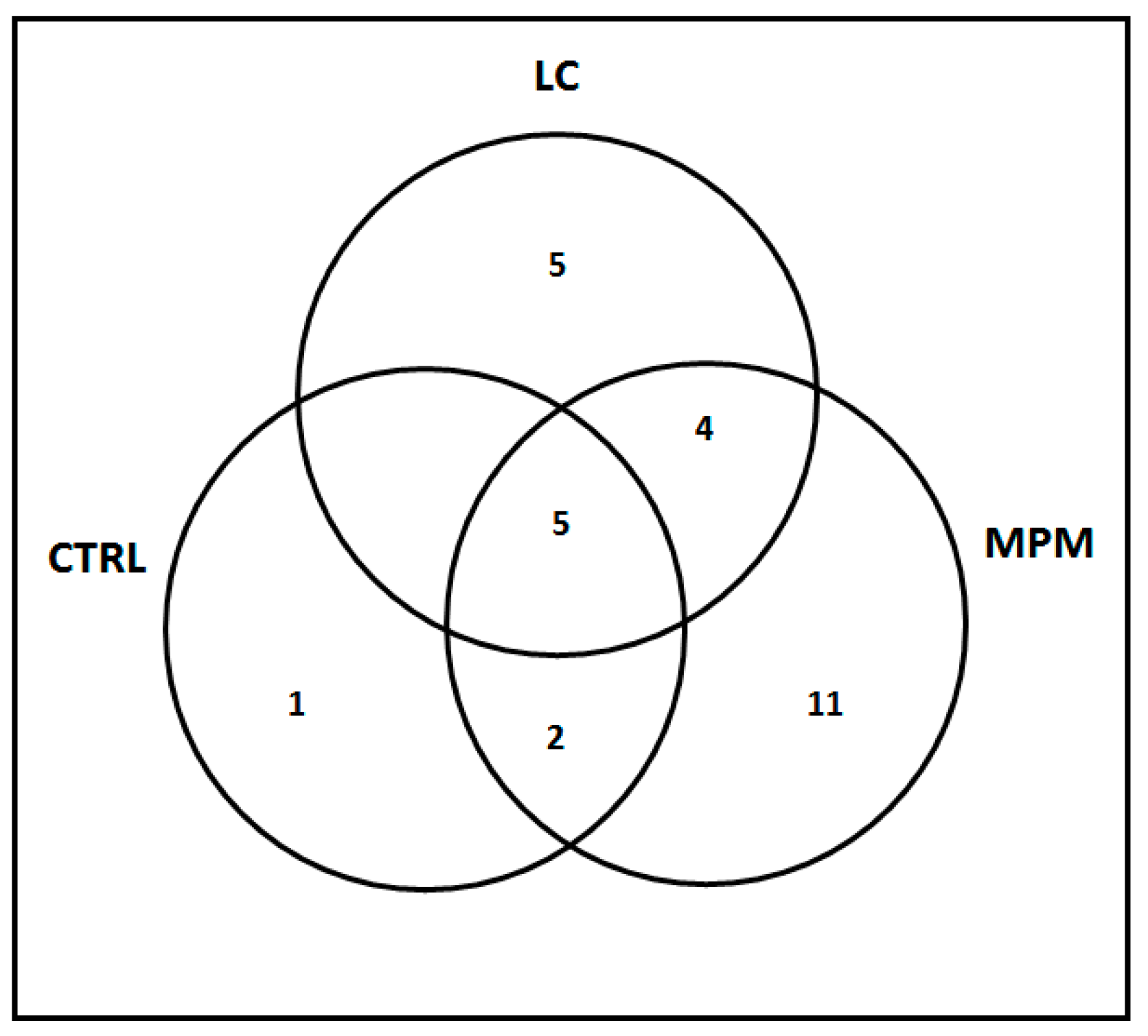
2.2. Genetic Analysis
2.3. Exclusive Enriched Pathways (EeEP)
- A tetrasaccharide linker sequence is required for glycosaminoglycans (GAG) synthesis (R-HSA-1971475);
- HS-GAG degradation (R-HSA-2024096);
- Defective beta-1,4-Galactosyltransferase 7 (B4GALT7) causes Ehlers–Danlos syndrome (EDS), progeroid type (R-HSA-3560783);
- Defective galactosylgalactosylxylosylprotein 3-beta-glucuronosyltransferases 3 (B3GAT3) causes joint dislocations, short stature, craniofacial dysmorphism, and congenital heart defects (JDSSDHD) (R-HSA-3560801);
- Defective -beta-1,3-galactosyltransferase 6 (B3GALT6) causes EDSP2 and spondyloepimetaphyseal dysplasia with joint laxity type 1 (SEMDJL1) (R-HSA-4420332).
- Beta Klotho-mediated ligand binding (R-HSA-1307965);
- Degradation of the extracellular matrix (R-HSA-1474228);
- Scavenging of heme from plasma (R-HSA-2168880)
- Nuclear envelope (NE) reassembly (R-HSA-2995410);
- Laminin interactions (R-HSA-3000157);
- Non-integrin membrane–ECM interactions (R-HSA-3000171);
- Defective Solute Carrier Family 29 Member 3 (SLC29A3) causes histiocytosis-lymphadenopathy plus syndrome (HLAS) (R-HSA-5619063);
- Infectious disease (R-HSA-5663205);
- Keratinization (R-HSA-6805567);
- Runt-related transcription factor 2 (RUNX2) regulates bone development (R-HSA-8941326);
- Interferon alpha/beta signaling (R-HSA-909733).
2.4. Shared Enriched Pathways (SeEP)
3. Discussion
3.1. VEA Applied to Thoracic Cancers Induced by Asbestos Exposure
3.1.1. Exclusive Enriched Pathways (eEP) for Controls (CTRL)
3.1.2. Exclusive Enriched Pathways (eEP) for LC
3.1.3. Exclusive Enriched Pathways (eEP) for MPM
Epithelial–Mesenchymal Transition (EMT) as the First Step in MPM Tumorigenesis (Figure 2, Point ➋)
The ECM Shaping of a Pro-Tumor Microenvironment (Figure 2, Point ➊)
Asbestos Fibers and the Nuclear Envelope (Figure 2, Point ➌)
The Emerging Role of Free Heme in MPM Pathogenesis (Figure 2, Point ➍)
3.1.4. The Possible Involvement of Retinol Metabolism and Transport and the Susceptibility to Develop MPM and LC (Figure 2, Point ➎)
3.2. Strength and Limitation of the Study
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. FFPE DNA Extraction
4.3. Exome Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andujar, P.; Lacourt, A.; Brochard, P.; Pairon, J.C.; Jaurand, M.C.; Jean, D. Five years update on relationships between malignant pleural mesothelioma and exposure to asbestos and other elongated mineral particles. J. Toxicol. Environ. Health B Crit. Rev. 2016, 19, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Gilham, C.; Rake, C.; Burdett, G.; Nicholson, A.G.; Davison, L.; Franchini, A.; Carpenter, J.; Hodgson, J.; Darnton, A.; Peto, J. Pleural mesothelioma and lung cancer risks in relation to occupational history and asbestos lung burden. Occup. Environ. Med. 2016, 73, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemen, R.A. Mesothelioma from asbestos exposures: Epidemiologic patterns and impact in the United States. J. Toxicol. Environ. Health B Crit. Rev. 2016, 19, 250–265. [Google Scholar] [CrossRef]
- Mossman, B.T. In vitro studies on the biologic effects of fibers: Correlation with in vivo bioassays. Environ. Health Perspect. 1990, 88, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Kamp, D.W.; Weitzman, S.A. The molecular basis of asbestos induced lung injury. Thorax 1999, 54, 638–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, S.J.; Daly, A.K.; Fairbrother, K.S.; Holmes, C.; Carpenter, C.L.; Navidi, W.C.; Idle, J.R. Lung cancer risk in African-Americans in relation to a race-specific CYP1A1 polymorphism. Cancer Res. 1995, 55, 6035–6037. [Google Scholar] [PubMed]
- Hirvonen, A.; Saarikoski, S.T.; Linnainmaa, K.; Koskinen, K.; Husgafvel-Pursiainen, K.; Mattson, K.; Vainio, H. Glutathione S-transferase and N-acetyltransferase genotypes and asbestos-associated pulmonary disorders. J. Natl. Cancer Inst. 1996, 88, 1853–1856. [Google Scholar] [CrossRef] [PubMed]
- Christiani, D.C. Smoking and the molecular epidemiology of lung cancer. Clin. Chest Med. 2000, 21, 87–93. [Google Scholar] [CrossRef]
- Schabath, M.B.; Spitz, M.R.; Delclos, G.L.; Gunn, G.B.; Whitehead, L.W.; Wu, X. Association between asbestos exposure, cigarette smoking, myeloperoxidase (MPO) genotypes, and lung cancer risk. Am. J. Ind. Med. 2002, 42, 29–37. [Google Scholar] [CrossRef]
- Wang, L.I.; Neuberg, D.; Christiani, D.C. Asbestos exposure, manganese superoxide dismutase (MnSOD) genotype, and lung cancer risk. J. Occup. Environ. Med. 2004, 46, 556–564. [Google Scholar] [CrossRef]
- Dianzani, I.; Gibello, L.; Biava, A.; Giordano, M.; Bertolotti, M.; Betti, M.; Ferrante, D.; Guarrera, S.; Betta, G.P.; Mirabelli, D.; et al. Polymorphisms in DNA repair genes as risk factors for asbestos-related malignant mesothelioma in a general population study. Mutat. Res. 2006, 599, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Landi, S.; Gemignani, F.; Neri, M.; Barale, R.; Bonassi, S.; Bottari, F.; Canessa, P.A.; Canzian, F.; Ceppi, M.; Filiberti, R.; et al. Polymorphisms of glutathione-S-transferase M1 and manganese superoxide dismutase. Int. J. Cancer 2007, 120, 2739–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugolini, D.; Neri, M.; Ceppi, M.; Cesario, A.; Dianzani, I.; Filiberti, R.; Gemignani, F.; Landi, S.; Magnani, C.; Mutti, L.; et al. Genetic susceptibility to malignant pleural mesothelioma and other asbestos-associated diseases. Mutat. Res. 2008, 659, 126–136. [Google Scholar]
- Gemignani, F.; Neri, M.; Bottari, F.; Barale, R.; Canessa, P.A.; Canzian, F.; Ceppi, M.; Spitaleri, I.; Cipollini, M.; Ivaldi, G.P.; et al. Risk of malignant pleural mesothelioma and polymorphisms in genes involved in the genome stability and xenobiotics metabolism. Mutat. Res. 2009, 671, 76–83. [Google Scholar] [CrossRef]
- Schneider, J.; Bernges, U. CYP1A1 and CYP1B1 polymorphisms as modifying factors in patients with pneumoconiosis and occupationally related tumours: A pilot study. Mol. Med. Rep. 2009, 2, 1023–1028. [Google Scholar] [CrossRef] [Green Version]
- Girardelli, M.; Maestri, I.; Rinaldi, R.R.; Tognon, M.; Boldorini, R.; Bovenzi, M.; Crovella, S.; Comar, M. NLRP1 polymorphisms in patients with asbestos-associated mesothelioma. Infect. Agents Cancer 2012, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Murakami, A.; Fujimori, Y.; Yoshikawa, Y.; Yamada, S.; Tamura, K.; Hirayama, N.; Terada, T.; Kuribayashi, K.; Tabata, C.; Fukuoka, K.; et al. Heme oxygenase-1 promoter polymorphism is associated with risk of malignant mesothelioma. Lung 2012, 190, 333–337. [Google Scholar] [CrossRef]
- Borelli, V.; Moura, R.R.; Trevisan, E.; Crovella, S. NLRP1 and NLRP3 polymorphisms in mesothelioma patients and asbestos exposed individuals a population-based autopsy study from North East Italy. Infect. Agents Cancer 2015, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Tunesi, S.; Ferrante, D.; Mirabelli, D.; Andorno, S.; Betti, M.; Fiorito, G.; Guarrera, S.; Casalone, E.; Neri, M.; Ugolini, D.; et al. Gene-asbestos interaction in malignant pleural mesothelioma susceptibility. Carcinogenesis 2015, 36, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Crovella, S.; Bianco, A.M.; Vuch, J.; Zupin, L.; Moura, R.R.; Trevisan, E.; Schneider, M.; Brollo, A.; Nicastro, E.M.; Cosenzi, A.; et al. Iron signature in asbestos-induced malignant pleural mesothelioma: A population-based autopsy study. J. Toxicol. Environ. Health A 2016, 79, 129–141. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Ferrante, D.; Sculco, M.; Righi, L.; Mirabelli, D.; Napoli, F.; Rondón-Lagos, M.; Casalone, E.; Vignolo Lutati, F.; et al. Sensitivity to asbestos is increased in patients with mesothelioma and pathogenic germline variants in BAP1 or other DNA repair genes. Genes Chromosomes Cancer 2018, 57, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Crovella, S.; Moura, R.R.; Cappellani, S.; Celsi, F.; Trevisan, E.; Schneider, M.; Brollo, A.; Nicastro, E.M.; Vita, F.; Finotto, L.; et al. A genetic variant of NLRP1 gene is associated with asbestos body burden in patients with malignant pleural mesothelioma. J. Toxicol. Environ. Health A 2018, 81, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wang, L.E.; McHugh, M.K.; Han, Y.; Xiong, M.; Amos, C.I.; Spitz, M.R.; Wei, Q.W. Genome-wide gene-environment interaction analysis for asbestos exposure in lung cancer susceptibility. Carcinogenesis 2012, 33, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadby, G.; Mukherjee, S.; Musk, A.W.; Reid, A.; Garlepp, M.; Dick, I.; Robinson, C.; Hui, J.; Fiorito, G.; Guarrera, S.; et al. A genome-wide association study for malignant mesothelioma risk. Lung Cancer 2013, 82, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Matullo, G.; Guarrera, S.; Betti, M.; Fiorito, G.; Ferrante, D.; Voglino, F.; Cadby, G.; Di Gaetano, C.; Rosa, F.; Russo, A.; et al. Genetic variants associated with increased risk of malignant pleural mesothelioma: A genome-wide association study. PLoS ONE 2013, 8, e61253. [Google Scholar] [CrossRef] [Green Version]
- Kettunen, E.; Hernandez-Vargas, H.; Cros, M.P.; Durand, G.; Le Calvez-Kelm, F.; Stuopelyte, K.; Jarmalaite, S.; Salmenkivi, K.; Anttila, S.; Wolff, H.; et al. Asbestos-associated genome-wide DNA methylation changes in lung cancer. Int. J. Cancer 2017, 141, 2014–2029. [Google Scholar] [CrossRef] [Green Version]
- Behrouzfar, K.; Burton, K.; Mutsaers, S.E.; Morahan, G.; Lake, R.A.; Fisher, S.A. How to Better Understand the Influence of Host Genetics on Developing an Effective Immune Response to Thoracic Cancers. Front. Oncol. 2021, 11, 679609. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 Mutations Predispose to Malignant Mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Ferris, L.K.; Baumann, F.; Napolitano, A.; Lum, C.A.; Flores, E.G.; Gaudino, G.; Powers, A.; Bryant-Greenwood, P.; Krausz, T.; et al. BAP1 Cancer Syndrome: Malignant Mesothelioma, Uveal and Cutaneous Melanoma, and Mbaits. J. Trans. Med. 2012, 10, 179. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Stücker, I.; Chen, C.; Goodman, G.; McHugh, M.K.; D’Amelio, A.M., Jr.; Etzel, C.J.; Li, S.; Lin, X.; Christiani, D.C. Genome-wide Gene-Asbestos Exposure Interaction Association Study Identifies a Common Susceptibility Variant on 22q13.31 Associated with Lung Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Brandão, L.A.C.; Moura, R.R.; Marzano, A.V.; Moltrasio, C.; Tricarico, P.M.; Crovella, S. Variant Enrichment Analysis to Explore Pathways Functionality in Complex Autoinflammatory Skin Disorders through Whole Exome Sequencing Analysis. Int. J. Mol. Sci. 2022, 23, 2278. [Google Scholar] [CrossRef] [PubMed]
- Crovella, S.; Revelant, A.; Muraro, E.; Moura, R.R.; Brandão, L.; Trovò, M.; Steffan, A.; Zacchi, P.; Zabucchi, G.; Minatel, E.; et al. Biological Pathways Associated with the Development of Pulmonary Toxicities in Mesothelioma Patients Treated with Radical Hemithoracic Radiation Therapy: A Preliminary Study. Front. Oncol. 2021, 11, 784081. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Brollo, A.; Ramani, L.; Bianchi, T.; Giarelli, L. Asbestos exposure in malignant mesothelioma of the pleura: A survey of 557 cases. Ind. Health 2001, 39, 161–167. [Google Scholar] [CrossRef]
- Casali, M.; Carugno, M.; Cattaneo, A.; Consonni, D.; Mensi, C.; Genovese, U.; Cavallo, D.M.; Somigliana, A.; Pesatori, A.C. Asbestos Lung Burden in Necroscopic Samples from the General Population of Milan, Italy. Ann. Occup. Hyg. 2015, 59, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Tossavainen, A. Asbestos, asbestosis, and cancer: The Helsinki criteria for diagnosis and attribution. Scand. J. Work Environ. Health 1997, 23, 311–316. [Google Scholar] [CrossRef]
- Klopfleisch, R.; Weiss, A.T.; Gruber, A.D. Excavation of a buried treasure--DNA, mRNA, miRNA and protein analysis in formalin fixed, paraffin embedded tissues. Histol. Histopathol. 2011, 26, 797–810. [Google Scholar]
- Solbes, E.; Harper, R.W. Biological responses to asbestos inhalation and pathogenesis of asbestos-related benign and malignant disease. J. Investig. Med. 2018, 66, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Blagoveshchenskaya, A.D.; Thomas, L.; Feliciangeli, S.F.; Hung, C.H.; Thomas, G. HIV-1 Nef Downregulates MHC-I by a PACS-1- and PI3K-Regulated ARF6 Endocytic Pathway. Cell 2002, 111, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Dikeakos, J.D.; Thomas, L.; Kwon, G.; Elferich, J.; Shinde, U.; Thomas, G. An interdomain binding site on HIV-1 Nef interacts with PACS-1 and PACS-2 on endosomes to down-regulate MHC-I. Mol. Biol. Cell 2012, 11, 2184–2197. [Google Scholar] [CrossRef]
- Thomas, G.; Aslan, J.E.; Thomas, L.; Shinde, P.; Shinde, U.; Simmen, T. Caught in the act—Protein adaptation and the expanding roles of the PACS proteins in tissue homeostasis and disease. J. Cell Sci. 2017, 130, 1865–1876. [Google Scholar] [CrossRef] [Green Version]
- Mani, C.; Tripathi, K.; Luan, S.; Clark, D.W.; Andrews, J.F.; Vindigni, A.; Thomas, G.; Palle, K. The multifunctional protein PACS-1 is required for HDAC2- and HDAC3-dependent chromatin maturation and genomic stability. Oncogene 2020, 39, 2583–2596. [Google Scholar] [CrossRef] [PubMed]
- Veena, M.S.; Raychaudhuri, S.; Basak, S.K.; Venkatesan, N.; Kumar, P.; Biswas, R.; Chakrabarti, R.; Lu, J.; Su, T.; Gallagher-Jones, M.; et al. Dysregulation of hsa-miR-34a and hsa-miR-449a leads to overexpression of PACS-1 and loss of DNA damage response (DDR) in cervical cancer. J. Biol. Chem. 2020, 295, 17169–17186. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.H.; Merry, C.L.; Davies, E.J.; Jayson, G.C. Heparan sulfate proteoglycans and cancer. Br. J. Cancer 2001, 85, 1094–1098. [Google Scholar] [CrossRef] [Green Version]
- De Pasquale, V.; Pavone, L.M. Heparan Sulfate Proteoglycan Signaling in Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 6588. [Google Scholar] [CrossRef]
- Lemjabbar-Alaoui, H.; van Zante, A.; Singer, M.S.; Xue, Q.; Wang, Y.Q.; Tsay, D.; He, B.; Jablons, D.M.; Rosen, S.D. Sulf-2, a heparan sulfate endosulfatase, promotes human lung carcinogenesis. Oncogene 2010, 29, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Jiang, J.; Shen, H.; Wang, H.; Zong, H.; Li, Z.; Yang, Y.; Niu, Z.; Liu, W.; Chen, X.; et al. Elevated beta-1,4-galactosyltransferase I in highly metastatic human lung cancer cells. Identification of E1AF as important transcription activator. J. Biol. Chem. 2005, 280, 12503–12516. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis. Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Repana, D.; Ross, P. Targeting FGF19/FGFR4 Pathway: A Novel Therapeutic Strategy for Hepatocellular Carcinoma. Diseases 2015, 3, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Liu, Y.; Qu, H. Expression of epithelial-mesenchymal transition-related genes increases with copy number in multiple cancer types. Oncotarget 2016, 7, 24688–24699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, M.; Cai, Y.; Li, X.; Zhao, C.; Cui, R. Dissecting the Role of the FGF19-FGFR4 Signaling Pathway in Cancer Development and Progression. Front. Cell Dev. Biol. 2020, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial–mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussos, E.T.; Keckesova, Z.; Haley, J.D.; Epstein, D.M.; Weinberg, R.A.; Condeelis, J.S. AACR special conference on epithelial-mesenchymal transition and cancer progression and treatment. Cancer Res. 2010, 70, 7360–7364. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial—Mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Qi, F.; Okimoto, G.; Jube, S.; Napolitano, A.; Pass, H.I.; Laczko, R.; Demay, R.M.; Khan, G.; Tiirikainen, M.; Rinaudo, C.; et al. Continuous exposure to chrysotile asbestos can cause transformation of human mesothelial cells via HMGB1 and TNF-α signaling. Am. J. Pathol. 2013, 183, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Turini, S.; Bergandi, L.; Gazzano, E.; Prato, M.; Aldieri, E. Epithelial to Mesenchymal Transition in Human Mesothelial Cells Exposed to Asbestos Fibers: Role of TGF-β as Mediator of Malignant Mesothelioma Development or Metastasis via EMT Event. Int. J. Mol. Sci. 2019, 20, 150. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.K.; MacPherson, M.B.; Beuschel, S.L.; Shukla, A. Asbestos-Induced Mesothelial to Fibroblastic Transition Is Modulated by the Inflammasome. Am. J. Pathol. 2017, 187, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Herreño, A.M.; Ramírez, A.C.; Chaparro, V.P.; Fernandez, M.J.; Cañas, A.; Morantes, C.F.; Moreno, O.M.; Brugés, R.E.; Mejía, J.A.; Bustos, F.J.; et al. Role of RUNX2 transcription factor in epithelial mesenchymal transition in non-small cell lung cancer lung cancer: Epigenetic control of the RUNX2 P1 promoter. Tumour Biol. 2019, 41, 1010428319851014. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Nasu, M.; Carbone, M.; Gaudino, G.; Ly, B.H.; Bertino, P.; Shimizu, D.; Morris, P.; Pass, H.I.; Yang, H. Ranpirnase Interferes with NF-κB Pathway and MMP9 Activity, Inhibiting Malignant Mesothelioma Cell Invasiveness and Xenograft Growth. Genes Cancer 2011, 2, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Wang, Y.; Lyu, L. Potential Roles of Matrix Metalloproteinases in Malignant Mesothelioma. In Asbestos-Related Diseases; Otsuki, T., Ed.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Simeone, P.; Trerotola, M.; Franck, J.; Cardon, T.; Marchisio, M.; Fournier, I.; Salzet, M.; Maffia, M.; Vergara, D. The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Biol. 2019, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Štrbac, D.; Dolžan, V. Matrix Metalloproteinases as Biomarkers and Treatment Targets in Mesothelioma: A Systematic Review. Biomolecules 2021, 11, 1272. [Google Scholar] [CrossRef] [PubMed]
- Štrbac, D.; Goričar, K.; Dolžan, V.; Kovač, V. Matrix Metalloproteinases Polymorphisms as Prognostic Biomarkers in Malignant Pleural Mesothelioma. Dis. Markers 2017, 2017, 8069529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennard, S.I.; Jaurand, M.C.; Bignon, J.; Kawanami, O.; Ferrans, V.J.; Davidson, J.; Crystal, R.G. Role of pleural mesothelial cells in the production of the submesothelial connective tissue matrix of lung. Am. Rev. Respir. Dis. 1984, 130, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.F.; Rinaldi, N.; Brüderlein, S.; Mechtersheimer, G.; Sträter, J.; Altevogt, P.; Möller, P. Mesothelial cells in suspension expose an enriched integrin repertoire capable of capturing soluble fibronectin and laminin. Cell Commun. Adhes. 2002, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.J.; Fang, C.C.; Chen, Y.M.; Lin, R.H.; Wu, K.D.; Lee, P.H.; Tsai, T.J. Extracellular matrix proteins modulate human peritoneal mesothelial cell behavior. Nephron 1997, 75, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Cox, T.R. The Role of the ECM in Lung Cancer Dormancy and Outgrowth. Front. Oncol. 2020, 10, 1766. [Google Scholar] [CrossRef]
- Wandke, C.; Kutay, U. Enclosing chromatin: Reassembly of the nucleus after open mitosis. Cell 2013, 152, 1222–1225. [Google Scholar] [CrossRef] [Green Version]
- Rüttner, J.R.; Lang, A.B.; Gut, D.R.; Wydler, M.U. Morphological aspects of interactions between asbestos fibers and human mesothelial cell cytoskeleton. Expl. Cell Biol. 1987, 55, 285–294. [Google Scholar] [CrossRef]
- Jensen, C.G.; Jensen, L.C.; Rieder, C.L.; Cole, R.W.; Ault, J.G. Long crocidolite asbestos fibers cause polyploidy by sterically blocking cytokinesis. Carcinogenesis 1996, 17, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Macnab, G.; Harington, J.S. Haemolytic activity of asbestos and other mineral dusts. Nature 1967, 214, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, R.J.; Pundsack, F.L. Asbestos hemolysis. Environ. Res. 1970, 3, 1–13. [Google Scholar] [CrossRef]
- Harington, J.S.; Miller, K.; Macnab, G. Hemolysis by asbestos. Environ. Res. 1971, 4, 95–117. [Google Scholar] [CrossRef]
- Nagai, H.; Ishihara, T.; Lee, W.H.; Ohara, H.; Okazaki, Y.; Okawa, K.; Toyokuni, S. Asbestos surface provides a niche for oxidative emodification. Cancer Sci. 2011, 102, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, V.; Chiabrando, D.; Petrillo, S.; Bertino, F.; Tolosano, E. The Multifaceted Role of Heme in Cancer. Front. Oncol. 2020, 9, 1540. [Google Scholar] [CrossRef] [Green Version]
- Shannahan, J.H.; Ghio, A.J.; Schladweiler, M.C.; McGee, J.K.; Richards, J.H.; Gavett, S.H.; Kodavanti, U.P. The role of iron in Libby amphibole-induced acute lung injury and inflammation. Inhal. Toxicol. 2011, 23, 313–323. [Google Scholar] [CrossRef]
- Aggarwal, S.; Ahmad, I.; Lam, A.; Carlisle, M.A.; Li, C.; Wells, J.M.; Raju, S.V.; Athar, M.; Rowe, S.M.; Dransfield, M.T.; et al. Heme scavenging reduces pulmonary endoplasmic reticulum stress, fibrosis, and emphysema. JCI Insight 2018, 3, e120694. [Google Scholar] [CrossRef] [Green Version]
- Afanas’eva, I.S.; Spitsyn, V.A.; Tsurikova, G.V. Genetic polymorphism of haptoglobin and quantitative changes in its levels during exposure to asbestos. Genetika 1993, 29, 1895–1900. [Google Scholar]
- Munson, P.; Lam, Y.W.; MacPherson, M.; Beuschel, S.; Shukla, A. Mouse serum exosomal proteomic signature in response to asbestos exposure. J. Cell. Biochem. 2018, 119, 6266–6273. [Google Scholar] [CrossRef] [PubMed]
- Omenn, G.S.; Goodman, G.; Thornquist, M.; Grizzle, J.; Rosenstock, L.; Barnhart, S.; Balmes, J.; Cherniack, M.G.; Cullen, M.R.; Glass, A. The beta-carotene and retinol efficacy trial (CARET) for chemoprevention of lung cancer in high risk populations: Smokers and asbestos-exposed workers. Cancer Res. 1994, 54, 2038s–2043s. [Google Scholar] [PubMed]
- Chuwers, P.; Barnhart, S.; Blanc, P.; Brodkin, C.A.; Cullen, M.; Kelly, T.; Keogh, J.; Omenn, G.; Williams, J.; Balmes, J.R. The protective effect of beta-carotene and retinol on ventilatory function in an asbestos-exposed cohort. Am. J. Respir. Crit. Care Med. 1997, 155, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Meyskens, F.L., Jr.; Omenn, G.S.; Valanis, B.; Williams, J.H.J. The Beta-Carotene and Retinol Efficacy Trial: Incidence of Lung Cancer and Cardiovascular Disease Mortality during 6-Year Follow-up after Stopping β-Carotene and Retinol Supplements. Natl. Cancer Inst. 2004, 96, 1743–1750. [Google Scholar] [CrossRef] [Green Version]
- Alfonso, H.S.; Reid, A.; de Klerk, N.H.; Olsen, N.; Mina, R.; Ambrosini, G.L.; Beilby, J.; Berry, G.; Musk, B.A. Retinol supplementation and mesothelioma incidence in workers earlier exposed to blue asbestos (Crocidolite) at Wittenoom, Western Australia. Eur. J. Cancer Prev. 2010, 19, 355–359. [Google Scholar] [CrossRef]
- Celsi, F.; Crovella, S.; Moura, R.R.; Schneider, M.; Vita, F.; Finotto, L.; Zabucchi, G.; Zacchi, P.; Borelli, V. Pleural mesothelioma and lung cancer: The role of asbestos exposure and genetic variants in selected iron metabolism and inflammation genes. J. Toxicol. Environ. Health A 2019, 82, 1088–1102. [Google Scholar] [CrossRef]
- Bianchi, C.; Brollo, A.; Ramani, L.; Zuch, C. Asbestos-related mesothelioma in Monfalcone, Italy. Am. J. Ind. Med. 1993, 24, 149–160. [Google Scholar] [CrossRef]
- Bianchi, C.; Brollo, A.; Ramani, L.; Berté, R. Exposure to asbestos in Monfalcone, Italy. A necropsy-based study. IARC Sci. Publ. 1991, 112, 127–140. [Google Scholar]
- Barbieri, P.G.; Consonni, D.; Somigliana, A. Relationship between pleural plaques and biomarkers of cumulative asbestos dose. A necropsy study. Med. Lav. 2019, 110, 353–362. [Google Scholar]
- Smith, M.J.; Naylor, B. A method for extracting ferruginous bodies from sputum and pulmonary tissue. Am. J. Clin. Pathol. 1972, 58, 250–254. [Google Scholar] [CrossRef]
- Istituto Superiore di Sanità; Biofibre Working Group. Asbestos Bodies in Human Lung Tissue and Biological Fluids: Analytical Method and Photo Atlas; Rapporti ISTISAN 17/12; Italian National Institute of Health (ISS): Rome, Italy, 2017; Volume IV, p. 58. (In Italian) [Google Scholar]
- Barbieri, P.G.; Somigliana, A.; Chen, Y.; Consonni, D.; Vignola, R.; Finotto, L. Lung Asbestos Fibre Burden and Pleural Mesothelioma in Women with Non-occupational Exposure. Ann. Work Expo. Health 2020, 64, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Inai, K. Pathology of mesothelioma. Environ. Health Prev. Med. 2008, 13, 60–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, A.G.; Tsao, M.S.; Beasley, M.B.; Borczuk, A.C.; Brambilla, E.; Cooper, W.A.; Dacic, S.; Jain, D.; Kerr, K.M.; Lantuejoul, S.; et al. The 2021 WHO Classification of Lung Tumors: Impact of Advances Since 2015. J. Thorac. Oncol. 2022, 17, 362–387. [Google Scholar] [CrossRef] [PubMed]
- Bisel, H.F.; Wroblewski, F.; Ladue, J.S. Incidence and clinical manifestations of cardiac metastases. J. Am. Med. Assoc. 1953, 153, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]

 ) preserving genomic stability: the possible role of PACS proteins. Susceptibility assets (exclusive to individuals who developed asbestos-related thoracic cancers, indicated with the symbol
) preserving genomic stability: the possible role of PACS proteins. Susceptibility assets (exclusive to individuals who developed asbestos-related thoracic cancers, indicated with the symbol  ): ➊ECM shaping of the lung/pleural tumor microenvironment in determining dormancy or outgrowth: abnormal ECM dynamics lead to deregulated cell proliferation and invasion, resulting in pathological processes including cancer. ➋ EMT as the initial step in tumorigenesis; ➌ Asbestos fibers and the nuclear envelope. Dysregulation of the nuclear envelope reassembly and susceptibility of developing MPM: a confirmation of the role of the well-documented close interaction between asbestos and the nuclear envelope of mesothelial cells in the induction of chromosome rearrangements seen in MPM; ➍ The emerging role of free heme in MPM pathogenesis: defective scavenging of free heme released by asbestos-fibers-induced hemolysis could contribute to asbestos-induced oxidative stress; ➎ The retinoid (Vitamin A) risk: dysregulated retinoid metabolism in asbestos-exposed individuals could contribute to asbestos-induced thoracic cancer pathogenesis. The histological image is a representative microphotograph relative to a Hematoxylin-and-Eosin-stained section of non-tumor lung which includes both pleural and lung tissue with the sole purpose of acting as a background for the schematic representation. Meso = Mesothelium, Red Blood Cells = RBC, AF = Asbestos Fibers, ROS = Reactive Oxygen Species, ECM = Extracellular Matrix, HS-GAGs = Heparan Sulphate Glycosaminoglycans, HSPGs = Heparan Sulphate ProteoGlycans.
) preserving genomic stability: the possible role of PACS proteins. Susceptibility assets (exclusive to individuals who developed asbestos-related thoracic cancers, indicated with the symbol ): ➊ECM shaping of the lung/pleural tumor microenvironment in determining dormancy or outgrowth: abnormal ECM dynamics lead to deregulated cell proliferation and invasion, resulting in pathological processes including cancer. ➋ EMT as the initial step in tumorigenesis; ➌ Asbestos fibers and the nuclear envelope. Dysregulation of the nuclear envelope reassembly and susceptibility of developing MPM: a confirmation of the role of the well-documented close interaction between asbestos and the nuclear envelope of mesothelial cells in the induction of chromosome rearrangements seen in MPM; ➍ The emerging role of free heme in MPM pathogenesis: defective scavenging of free heme released by asbestos-fibers-induced hemolysis could contribute to asbestos-induced oxidative stress; ➎ The retinoid (Vitamin A) risk: dysregulated retinoid metabolism in asbestos-exposed individuals could contribute to asbestos-induced thoracic cancer pathogenesis. The histological image is a representative microphotograph relative to a Hematoxylin-and-Eosin-stained section of non-tumor lung which includes both pleural and lung tissue with the sole purpose of acting as a background for the schematic representation. Meso = Mesothelium, Red Blood Cells = RBC, AF = Asbestos Fibers, ROS = Reactive Oxygen Species, ECM = Extracellular Matrix, HS-GAGs = Heparan Sulphate Glycosaminoglycans, HSPGs = Heparan Sulphate ProteoGlycans.
): ➊ECM shaping of the lung/pleural tumor microenvironment in determining dormancy or outgrowth: abnormal ECM dynamics lead to deregulated cell proliferation and invasion, resulting in pathological processes including cancer. ➋ EMT as the initial step in tumorigenesis; ➌ Asbestos fibers and the nuclear envelope. Dysregulation of the nuclear envelope reassembly and susceptibility of developing MPM: a confirmation of the role of the well-documented close interaction between asbestos and the nuclear envelope of mesothelial cells in the induction of chromosome rearrangements seen in MPM; ➍ The emerging role of free heme in MPM pathogenesis: defective scavenging of free heme released by asbestos-fibers-induced hemolysis could contribute to asbestos-induced oxidative stress; ➎ The retinoid (Vitamin A) risk: dysregulated retinoid metabolism in asbestos-exposed individuals could contribute to asbestos-induced thoracic cancer pathogenesis. The histological image is a representative microphotograph relative to a Hematoxylin-and-Eosin-stained section of non-tumor lung which includes both pleural and lung tissue with the sole purpose of acting as a background for the schematic representation. Meso = Mesothelium, Red Blood Cells = RBC, AF = Asbestos Fibers, ROS = Reactive Oxygen Species, ECM = Extracellular Matrix, HS-GAGs = Heparan Sulphate Glycosaminoglycans, HSPGs = Heparan Sulphate ProteoGlycans.
) preserving genomic stability: the possible role of PACS proteins. Susceptibility assets (exclusive to individuals who developed asbestos-related thoracic cancers, indicated with the symbol ): ➊ECM shaping of the lung/pleural tumor microenvironment in determining dormancy or outgrowth: abnormal ECM dynamics lead to deregulated cell proliferation and invasion, resulting in pathological processes including cancer. ➋ EMT as the initial step in tumorigenesis; ➌ Asbestos fibers and the nuclear envelope. Dysregulation of the nuclear envelope reassembly and susceptibility of developing MPM: a confirmation of the role of the well-documented close interaction between asbestos and the nuclear envelope of mesothelial cells in the induction of chromosome rearrangements seen in MPM; ➍ The emerging role of free heme in MPM pathogenesis: defective scavenging of free heme released by asbestos-fibers-induced hemolysis could contribute to asbestos-induced oxidative stress; ➎ The retinoid (Vitamin A) risk: dysregulated retinoid metabolism in asbestos-exposed individuals could contribute to asbestos-induced thoracic cancer pathogenesis. The histological image is a representative microphotograph relative to a Hematoxylin-and-Eosin-stained section of non-tumor lung which includes both pleural and lung tissue with the sole purpose of acting as a background for the schematic representation. Meso = Mesothelium, Red Blood Cells = RBC, AF = Asbestos Fibers, ROS = Reactive Oxygen Species, ECM = Extracellular Matrix, HS-GAGs = Heparan Sulphate Glycosaminoglycans, HSPGs = Heparan Sulphate ProteoGlycans.

{kind=link}
{kind=link}
| CTRL | LC | MPM | |
|---|---|---|---|
| Number of cases | 5 | 7 | 7 |
| Mean age (years ± sd) | 79.4 ± 2.2 | 76.1 ± 8.6 | 76.9 ± 6.3 |
| Cause of death | Cardiovascular diseases | SCLC [2]; NSCLC [5] | EMPM [2]; BMPM [2]; SMPM [3] |
| Asbestos bodies count, in n/g dry tissue, (mean ± sd) | 1.30 × 105 (6.07 × 104) | 1.02 × 105 (1.48 × 105) | 1.27 × 105 (9.90 × 104) |
| Asbestos fibers count, in n/g dry tissue, (mean ± sd) | NA | 1.04 × 107 (1.37 × 107) | 1.86 × 107 (2.72 × 107) |
| Number of hyaline plaques | |||
| Absent | 0 | 0 | 0 |
| Grade 1 | 0 | 0 | 1 |
| Grade 2 | 1 | 6 | 3 |
| Grade 3 | 4 | 1 | 3 |
| Group | Reactome ID | Reactome Pathway Name | VariantRatio | BgRatio | OR | CI95− | CI95+ | adj. p-Value |
|---|---|---|---|---|---|---|---|---|
| CTRL | R-HSA-164940 | Nef-mediated downregulation of MHC class I complex cell surface expression | 12/5610 | 69/180,208 | 5.59 | 2.75 | 10.40 | 1.43 × 10−2 |
| R-HSA-2172127 | DAP12 interactions | 26/5610 | 311/180,208 | 2.69 | 1.73 | 4.02 | 4.02 × 10−2 | |
| R-HSA-5083632 | Defective C1GALT1C1 causes Tn polyagglutination syndrome (TNPS) | 148/5610 | 1809/180,208 | 2.63 | 2.20 | 3.12 | 1.42 × 10−19 | |
| R-HSA-977068 | Termination of O-glycan biosynthesis | 151/5610 | 1847/180,208 | 2.63 | 2.21 | 3.11 | 5.57 × 10−20 | |
| R-HSA-5083625 | Defective GALNT3 causes familial hyperphosphatemic tumoral calcinosis (HFTC) | 148/5610 | 1811/180,208 | 2.63 | 2.20 | 3.11 | 1.55 × 10−19 | |
| R-HSA-5083636 | Defective GALNT12 causes colorectal cancer 1 (CRCS1) | 148/5610 | 1812/180,208 | 2.62 | 2.20 | 3.11 | 1.61 × 10−19 | |
| R-HSA-5621480 | Dectin-2 family | 151/5610 | 1892/180,208 | 2.56 | 2.15 | 3.03 | 4.49 × 10−19 | |
| R-HSA-198933 | Immunoregulatory interactions between a lymphoid and a non-lymphoid cell | 99/5610 | 1750/180,208 | 1.82 | 1.47 | 2.23 | 3.00 × 10−4 | |
| LC | R-HSA-1839128 | FGFR4 mutant receptor activation | 6/4102 | 16/180,208 | 16.48 | 5.28 | 44.31 | 1.63 × 10−2 |
| R-HSA-3656237 | Defective EXT2 causes exostoses 2 | 20/4102 | 211/180,208 | 4.16 | 2.49 | 6.60 | 8.21 × 10−4 | |
| R-HSA-3656253 | Defective EXT1 causes exostoses 1, TRPS2 and CHDS | 20/4102 | 211/180,208 | 4.16 | 2.49 | 6.60 | 8.21 × 10−4 | |
| R-HSA-2024096 | HS-GAG degradation | 25/4102 | 312/180,208 | 3.52 | 2.24 | 5.30 | 6.02 × 10−4 | |
| R-HSA-3560801 | Defective B3GAT3 causes JDSSDHD | 21/4102 | 270/180,208 | 3.42 | 2.08 | 5.34 | 8.07 × 10−3 | |
| R-HSA-4420332 | Defective B3GALT6 causes EDSP2 and SEMDJL1 | 20/4102 | 266/180,208 | 3.30 | 1.98 | 5.21 | 2.17 × 10−2 | |
| R-HSA-3560783 | Defective B4GALT7 causes EDS, progeroid type | 20/4102 | 269/180,208 | 3.27 | 1.96 | 5.15 | 2.52 × 10−2 | |
| R-HSA-1971475 | A tetrasaccharide linker sequence is required for GAG synthesis | 21/4102 | 304/180,208 | 3.03 | 1.85 | 4.73 | 4.33 × 10−2 | |
| R-HSA-975634 | Retinoid metabolism and transport | 32/4102 | 571/180,208 | 2.46 | 1.67 | 3.52 | 2.48 × 10−2 | |
| R-HSA-977068 | Termination of O-glycan biosynthesis | 93/4102 | 1847/180,208 | 2.21 | 1.77 | 2.73 | 6.91 × 10−8 | |
| R-HSA-5083632 | Defective C1GALT1C1 causes Tn polyagglutination syndrome (TNPS) | 91/4102 | 1809/180,208 | 2.21 | 1.77 | 2.74 | 1.22 × 10−7 | |
| R-HSA-5083625 | Defective GALNT3 causes familial hyperphosphatemic tumoral calcinosis (HFTC) | 91/4102 | 1811/180,208 | 2.21 | 1.76 | 2.73 | 1.26 × 10−7 | |
| R-HSA-5083636 | Defective GALNT12 causes colorectal cancer 1 (CRCS1) | 91/4102 | 1812/180,208 | 2.21 | 1.76 | 2.73 | 1.29 × 10−7 | |
| R-HSA-5621480 | Dectin-2 family | 92/4102 | 1892/180,208 | 2.14 | 1.71 | 2.64 | 5.42 × 10−7 | |
| MPM | R-HSA-5619063 | Defective SLC29A3 causes histiocytosis-lymphadenopathy plus syndrome (HLAS) | 6/7745 | 9/180,208 | 15.51 | 4.54 | 48.84 | 4.32 × 10−2 |
| R-HSA-1839128 | FGFR4 mutant receptor activation | 9/7745 | 16/180,208 | 13.09 | 5.10 | 31.46 | 9.35 × 10−4 | |
| R-HSA-1307965 | betaKlotho-mediated ligand binding | 9/7745 | 21/180,208 | 9.97 | 4.02 | 22.72 | 5.43 × 10−3 | |
| R-HSA-2172127 | DAP12 interactions | 57/7745 | 311/180,208 | 4.26 | 3.15 | 5.68 | 3.79 × 10−14 | |
| R-HSA-2995410 | Nuclear envelope (NE) reassembly | 17/7745 | 93/180,208 | 4.25 | 2.38 | 7.19 | 6.90 × 10−3 | |
| R-HSA-2168880 | Scavenging of heme from plasma | 17/7745 | 108/180,208 | 3.66 | 2.06 | 6.14 | 3.97 × 10−2 | |
| R-HSA-3656237 | Defective EXT2 causes exostoses 2 | 26/7745 | 211/180,208 | 2.87 | 1.83 | 4.32 | 1.66 × 10−2 | |
| R-HSA-3656253 | Defective EXT1 causes exostoses 1, TRPS2 and CHDS | 26/7745 | 211/180,208 | 2.87 | 1.83 | 4.32 | 1.66 × 10−2 | |
| R-HSA-8941326 | RUNX2 regulates bone development | 28/7745 | 234/180,208 | 2.78 | 1.81 | 4.13 | 1.25 × 10−2 | |
| R-HSA-5663205 | Infectious disease | 256/7745 | 2720/180,208 | 2.19 | 1.92 | 2.50 | 2.76 × 10−23 | |
| R-HSA-977068 | Termination of O-glycan biosynthesis | 170/7745 | 1847/180,208 | 2.14 | 1.82 | 2.51 | 3.42 × 10−14 | |
| R-HSA-5083632 | Defective C1GALT1C1 causes Tn polyagglutination syndrome (TNPS) | 166/7745 | 1809/180,208 | 2.14 | 1.81 | 2.51 | 1.12 × 10−13 | |
| R-HSA-5083625 | Defective GALNT3 causes familial hyperphosphatemic tumoral calcinosis (HFTC) | 166/7745 | 1811/180,208 | 2.13 | 1.81 | 2.51 | 1.17 × 10−13 | |
| R-HSA-5083636 | Defective GALNT12 causes colorectal cancer 1 (CRCS1) | 166/7745 | 1812/180,208 | 2.13 | 1.80 | 2.50 | 1.20 × 10−13 | |
| R-HSA-975634 | Retinoid metabolism and transport | 51/7745 | 571/180,208 | 2.08 | 1.53 | 2.77 | 1.56 × 10−2 | |
| R-HSA-5621480 | Dectin-2 family | 168/7745 | 1892/180,208 | 2.07 | 1.75 | 2.42 | 9.39 × 10−13 | |
| R-HSA-3000157 | Laminin interactions | 79/7745 | 988/180,208 | 1.86 | 1.46 | 2.34 | 2.36 × 10−3 | |
| R-HSA-909733 | Interferon alpha/beta signaling | 79/7745 | 1010/180,208 | 1.82 | 1.43 | 2.29 | 6.34 × 10−3 | |
| R-HSA-198933 | Immunoregulatory interactions between a lymphoid and a non-lymphoid cell | 131/7745 | 1750/180,208 | 1.74 | 1.45 | 2.08 | 3.45 × 10−5 | |
| R-HSA-3000171 | Non-integrin membrane–ECM interactions | 108/7745 | 1556/180,208 | 1.61 | 1.31 | 1.97 | 1.86 × 10−2 | |
| R-HSA-6805567 | Keratinization | 108/7745 | 1563/180,208 | 1.61 | 1.31 | 1.96 | 1.97 × 10−2 | |
| R-HSA-1474228 | Degradation of the extracellular matrix | 185/7745 | 2814/180,208 | 1.53 | 1.31 | 1.78 | 4.04 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crovella, S.; Moura, R.R.; Brandão, L.; Vita, F.; Schneider, M.; Zanconati, F.; Finotto, L.; Zacchi, P.; Zabucchi, G.; Borelli, V. Variant Enrichment Analysis to Explore Pathways Disruption in a Necropsy Series of Asbestos-Exposed Shipyard Workers. Int. J. Mol. Sci. 2022, 23, 13628. https://doi.org/10.3390/ijms232113628
Crovella S, Moura RR, Brandão L, Vita F, Schneider M, Zanconati F, Finotto L, Zacchi P, Zabucchi G, Borelli V. Variant Enrichment Analysis to Explore Pathways Disruption in a Necropsy Series of Asbestos-Exposed Shipyard Workers. International Journal of Molecular Sciences. 2022; 23(21):13628. https://doi.org/10.3390/ijms232113628
Chicago/Turabian StyleCrovella, Sergio, Ronald Rodrigues Moura, Lucas Brandão, Francesca Vita, Manuela Schneider, Fabrizio Zanconati, Luigi Finotto, Paola Zacchi, Giuliano Zabucchi, and Violetta Borelli. 2022. "Variant Enrichment Analysis to Explore Pathways Disruption in a Necropsy Series of Asbestos-Exposed Shipyard Workers" International Journal of Molecular Sciences 23, no. 21: 13628. https://doi.org/10.3390/ijms232113628