
Molecular Mechanisms of Ischaemia-Reperfusion Injury and Regeneration in the Liver-Shock and Surgery-Associated Changes
 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
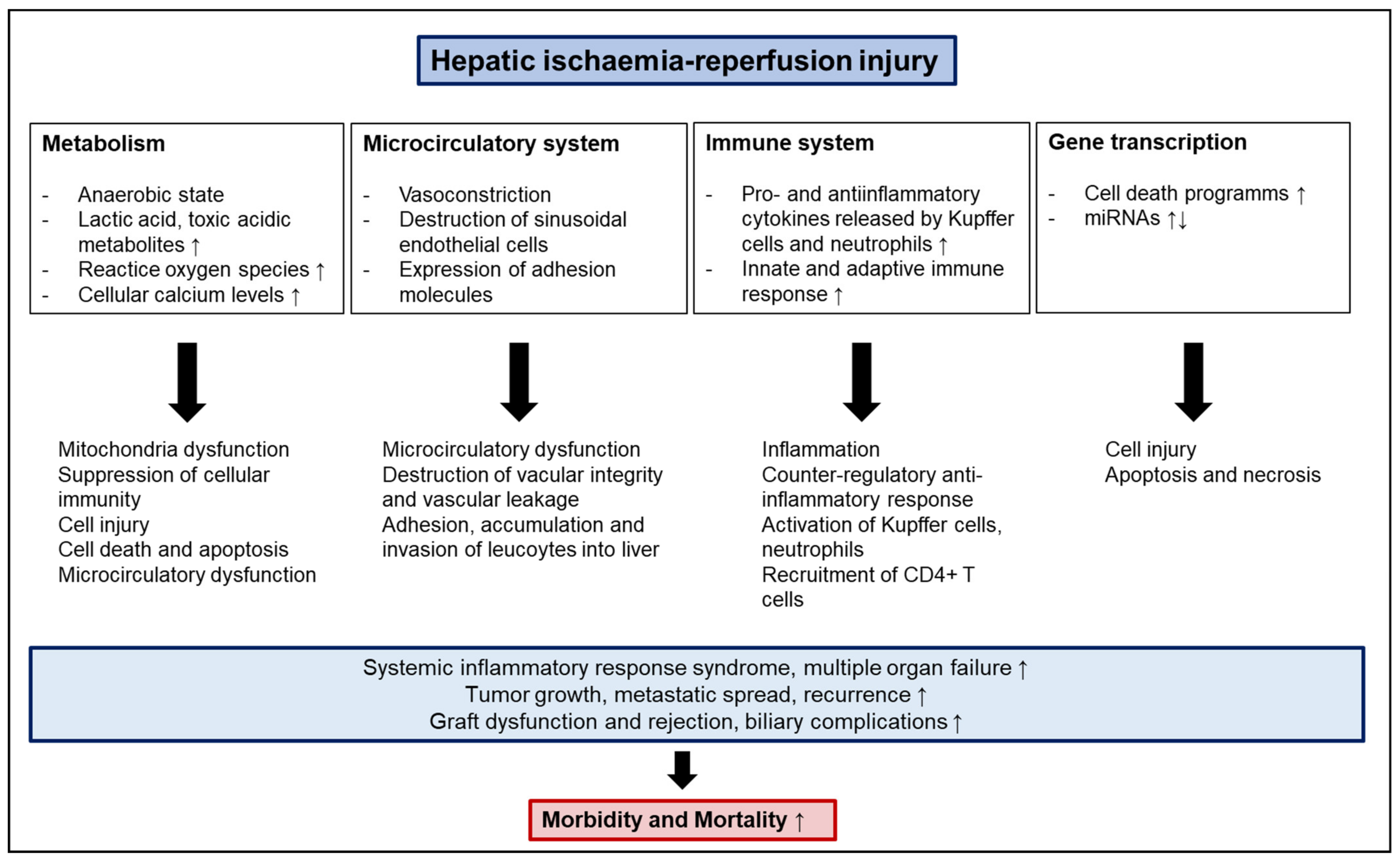
2. Molecular Mechanisms and Pathophysiology of IRI
2.1. Anaerobic Metabolism, Acidosis, and ATP Depletion
2.2. Mitochondria, Oxidative Stress and Reactive Oxygen Species
2.3. Microcirculatory Dysfunction
2.4. Pro- and Anti-Inflammatory Cytokines
2.5. NF-κB
2.6. Apoptosis and Necrosis
2.7. miRNAs
2.8. Innate and Adaptive Immune Activation
3. Cold Ischemia Versus Warm Ischemia Reperfusion Injury
3.1. Cold Ischemia
3.2. Warm Ischemia
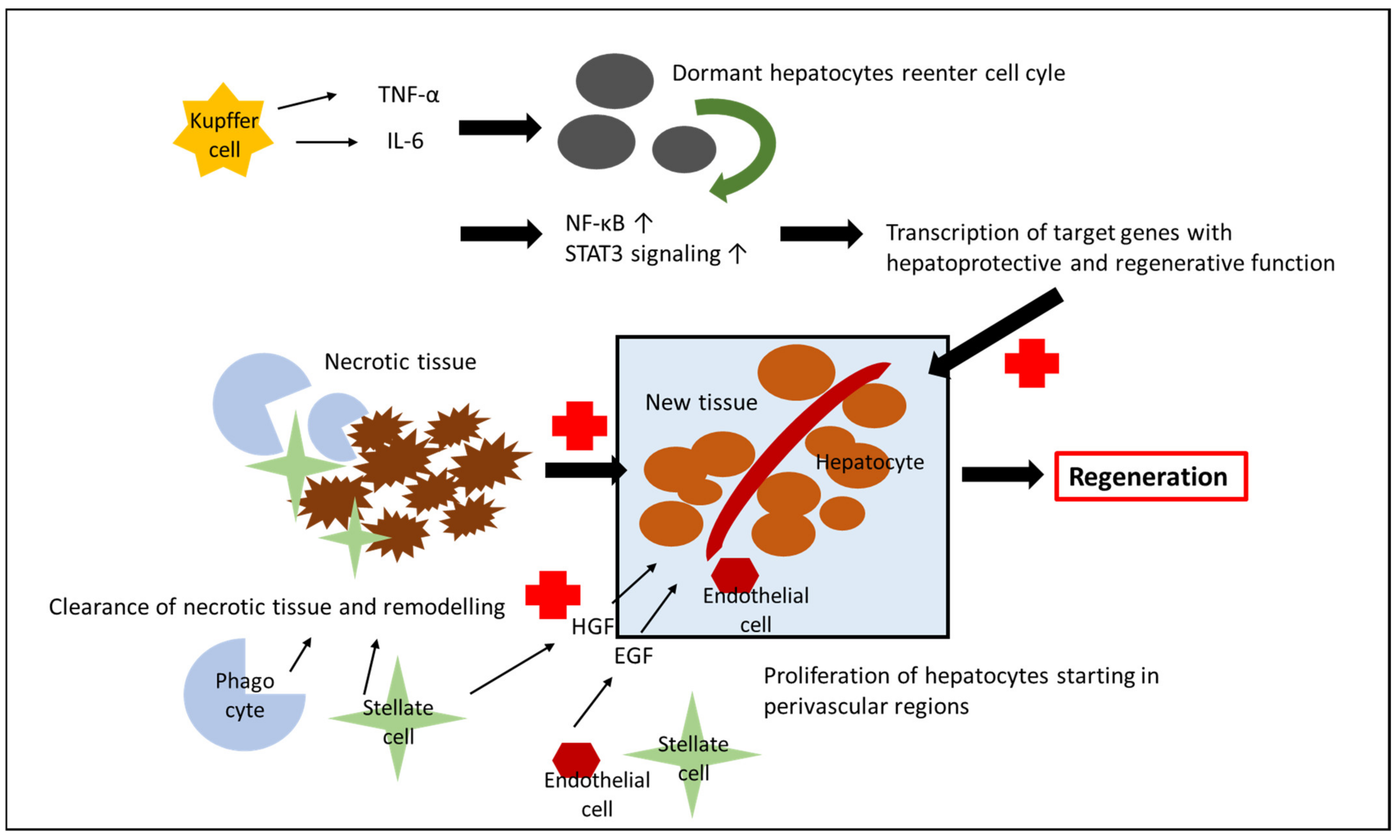
4. Liver Regeneration after IRI
5. Clinical Implications of IRI in Different Settings
5.1. Tumor Growth, Metastasis, and Recurrence
5.2. Graft Fibrosis
5.3. Biliary Complications and Graft Dysfunction
6. Therapeutic Approaches
7. Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Toledo-Pereyra, L.H.; Simmons, R.L.; Najarian, J.S. Protection of the ischemic liver by donor pretreatment before transplantation. Am. J. Surg. 1975, 129, 513–517. [Google Scholar] [CrossRef]
- Singer, M.; Young, P.J.; Laffey, J.G.; Asfar, P.; Taccone, F.S.; Skrifvars, M.B.; Meyhoff, C.S.; Radermacher, P. Dangers of hyperoxia. Crit. Care 2021, 25, 440. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.; Eckle, T. Ischemia and reperfusion-from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Nastos, C.; Kalimeris, K.; Papoutsidakis, N.; Tasoulis, M.K.; Lykoudis, P.M.; Theodoraki, K.; Nastou, D.; Smyrniotis, V.; Arkadopoulos, N. Global consequences of liver ischemia/reperfusion injury. Oxid. Med. Cell. Longev. 2014, 2014, 906965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serracino-Inglott, F.; Habib, N.A.; Mathie, R.T. Hepatic ischemia-reperfusion injury. Am. J. Surg. 2001, 181, 160–166. [Google Scholar] [CrossRef]
- Man, K.; Ng, K.; Mau Lo, C.; Ho, J.; Sun, B.; Sun, C.; Lee, T.; Poon, R.; Fan, S. Ischemia-Reperfusion of Small Liver Remnant Promotes Liver Tumor Growth and Metastases—Activation of Cell Invasion and Migration Pathways. Liver Transplant. 2007, 13, 767–768. [Google Scholar] [CrossRef]
- Pretzsch, E.; Bösch, F.; Renz, B.; Werner, J.; Angele, M.; Chaudry, I.H. Operative Trauma and Blood Loss—Impact on Tumor Growth and Recurrence. Shock 2021, 55, 455–464. [Google Scholar] [CrossRef]
- Kannerup, A.S.; Grønbæk, H.; Funch-Jensen, P.; Karlsen, S.; Mortensen, F.V. Partial liver ischemia is followed by metabolic changes in the normally perfused part of the liver during reperfusion. Eur. Surg. Res. 2010, 45, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G.; Ungerstedt, J.; Wernerman, J.; Ungerstedt, U.; Ericzon, B.G. Metabolic changes in the liver graft monitored continuously with microdialysis during liver transplantation in a pig model. Liver Transplant. 2002, 8, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yan, Q.; Wang, X.; Chen, X.; Chen, Y.; Du, J.; Chen, L. The Role of Mitochondria in Liver Ischemia-Reperfusion Injury: From Aspects of Mitochondrial Oxidative Stress, Mitochondrial Fission, Mitochondrial Membrane Permeable Transport Pore Formation, Mitophagy, and Mitochondria-Related Protective Measures. Oxid. Med. Cell. Longev. 2021, 2021, 6670579. [Google Scholar] [CrossRef]
- Cannistrà, M.; Ruggiero, M.; Zullo, A.; Gallelli, G.; Serafini, S.; Maria, M.; Naso, A.; Grande, R.; Serra, R.; Nardo, B. Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int. J. Surg. 2016, 33, S57–S70. [Google Scholar] [CrossRef] [PubMed]
- Chaudry, I.H.; Ayala, A. Mechanism of increased susceptibility to infection following hemorrhage. Am. J. Surg. 1993, 165, 59S–67S. [Google Scholar] [CrossRef]
- Ayala, A.; Perrin, M.; Ertel, W.; Chaudry, I. Differential effects of hemorrhage on kupffer cells: Decreased antigen presentation despite increased inflammatory cytokine (IL-1, IL-6 and TNF) release. Cytokine 1992, 4, 66–75. [Google Scholar] [CrossRef]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-Induced Apoptosis Through Calcineurin Dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [CrossRef]
- Datta, G.; Fuller, B.J.; Davidson, B.R. Molecular mechanisms of liver ischemia reperfusion injury: Insights from transgenic knockout models. World J. Gastroenterol. 2013, 19, 1683–1698. [Google Scholar] [CrossRef]
- Jaeschke, H. Reactive oxygen and ischemia/reperfusion injury of the liver. Chem. Biol. Interact. 1991, 79, 115–136. [Google Scholar] [CrossRef]
- Kis, A.; Yellon, D.M.; Baxter, G.F. Role of nuclear factor-κB activation in acute ischaemia-reperfusion injury in myocardium. Br. J. Pharmacol. 2003, 138, 894–900. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Rauen, U.; Polzar, B.; Stephan, H.; Mannherz, H.; De Groot, H. Cold-induced apoptosis in cultured hepatocytes and liver endothelial cells: Mediation by reactive oxygen species. FASEB J. 1999, 13, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankenberg, M.V.; Weimann, J.; Fritz, S.; Fiedler, J.; Mehrabi, A.; Büchler, M.W.; Kraus, T.W. Gadolinium chloride-induced improvement of postischemic hepatic perfusion after warm ischemia is associated with reduced hepatic endothelin secretion. Transpl. Int. 2005, 18, 429–436. [Google Scholar] [CrossRef]
- Uhlmann, D.; Gaebel, G.; Armann, B.; Ludwig, S.; Hess, J.; Pietsch, U.C.; Fiedler, M.; Tannapfel, A.; Hauss, J.; Witzigmann, H. Attenuation of proinflammatory gene expression and microcirculatory disturbances by endothelinA receptor blockade after orthotopic liver transplantation in pigs. Surgery 2006, 139, 61–72. [Google Scholar] [CrossRef]
- Peralta, C.; Fernández, L.; Panés, J.; Prats, N.; Sans, M.; Piqué, J.M.; Gelpí, E.; Roselló-Catafau, J. Preconditioning protects against systemic disorders associated with hepatic ischemia-reperfusion through blockade of tumor necrosis factor-induced P-selectin up-regulation in the rat. Hepatology 2001, 33, 100–113. [Google Scholar] [CrossRef]
- Siriussawakul, A.; Zaky, A.; Lang, J.D. Role of nitric oxide in hepatic ischemia-reperfusion injury. World J. Gastroenterol. 2010, 16, 6079–6086. [Google Scholar] [CrossRef]
- Burke, J.; Zibari, G.B.; Brown, M.F.; Granger, N.; Kelly, R.; Singh, I.; Mcdonald, J.C. Hepatic Ischemia–Reperfusion Injury Causes E-Selectin Upregulation. Transplant. Proc. 1998, 1345, 2321–2323. [Google Scholar] [CrossRef]
- Lentsch, A.B.; Yoshidome, H.; Cheadle, W.G.; Miller, F.N.; Edwards, M.J. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: Roles for macrophage inflammatory protein-2 and Kupffer cells. Hepatology 1998, 27, 507–512. [Google Scholar] [CrossRef]
- McKeown, C.M.B.; Edwards, V.; Phillips, M.J.; Harvey, P.R.; Petrunka, C.N.; Strasberg, S.M. Sinusoidal lining cell damage: The critical injury in cold preservation of liver allografts in the rat. Transplantation 1988, 46, 178–191. [Google Scholar] [CrossRef]
- Gujral, J.S.; Bucci, T.J.; Farhood, A.; Jaeschke, H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: Apoptosis or necrosis? Hepatology 2001, 33, 397–405. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of liver injury. I. TNF-α-induced liver injury: Role of IKK, JNK, and ROS pathways. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, 583–589. [Google Scholar] [CrossRef]
- Oe, S.; Hirose, T.; Fujii, H.; Yasuchika, K.; Nishio, T.; Iimuro, Y.; Morimoto, T.; Nagao, M.; Yamaoka, Y. Continuous intravenous infusion of deleted form of hepatocyte growth factor attenuates hepatic ischemia-reperfusion injury in rats. J. Hepatol. 2001, 34, 832–839. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty Acid and Endotoxin Activate Inflammasomes in Mouse Hepatocytes that Release Danger Signals to Stimulate Immune Cells. Hepatology 2012, 54, 133–144. [Google Scholar] [CrossRef]
- Kato, A.; Graul-Layman, A.; Edwards, M.; Lentsch, A. Promotion Of Hepatic Ischemia/Reperfusion Injury By ILl-12 Is Independent Of STAT4. Transplantation 2002, 73, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Klune, J.; Bartels, C.; Luo, J.; Yokota, S.; Du, Q.; Geller, D. IL-23 mediated murine liver transplantation reperfusion injury via IFNy/IRF-1 pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Teoh, N.; Field, J.; Farrell, G. Interleukin-6 is a key mediator of the hepatoprotective and pro-proliferative effects of ischaemic preconditioning in mice. J. Hepatol. 2006, 45, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Tsurui, Y.; Sho, M.; Kuzumoto, Y.; Hamada, K.; Akashi, S.; Kashizuka, H.; Ikeda, N.; Nomi, T.; Mizuno, T.; Kanehiro, H.; et al. Dual role of vascular endothelial growth factor in hepatic ischemia-reperfusion injury. Transplantation 2005, 79, 1110–1115. [Google Scholar] [CrossRef]
- Chen, X.H.; Minatoguchi, S.; Kosai, K.; Yuge, K.; Takahashi, T.; Arai, M.; Wang, N.; Misao, Y.; Lu, C.; Onogi, H.; et al. In Vivo Hepatocyte Growth Factor Gene Transfer Reduces Myocardial Ischemia-Reperfusion Injury Through Its Multiple Actions. J. Card. Fail. 2007, 13, 874–883. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, D.; Chen, S.; Li, X.; Ge, M.; Huang, W. Sevoflurane preconditioning activates HGF/Met-mediated autophagy to attenuate hepatic ischemia-reperfusion injury in mice. Cell. Signal. 2021, 82, 109966. [Google Scholar] [CrossRef]
- Konishi, T.; Lentsch, A.B. Hepatic ischemia/reperfusion: Mechanisms of tissue injury, repair, and regeneration. Gene Expr. 2017, 17, 277–287. [Google Scholar] [CrossRef]
- Angele, M.K.; Faist, E. Clinical review: Immunodepression in the surgical patient and increased susceptibility to infection. Crit. Care 2002, 6, 298–305. [Google Scholar] [CrossRef]
- Lin, E.; Calvano, S.E.; Lowry, S.F. Inflammatory cytokines and cell response in surgery. Surgery 2000, 127, 117–126. [Google Scholar] [CrossRef]
- Li, J.D.; Peng, Y.; Peng, X.Y.; Li, Q.L.; Li, Q. Suppression of Nuclear Factor-κB Activity in Kupffer Cells Protects Rat Liver Graft From Ischemia-Reperfusion Injury. Transplant. Proc. 2010, 42, 1582–1586. [Google Scholar] [CrossRef]
- Luedde, T.; Assmus, U.; Wüstefeld, T.; Meyer Zu Vilsendorf, A.; Roskams, T.; Schmidt-Supprian, M.; Rajewsky, K.; Brenner, D.A.; Manns, M.P.; Pasparakis, M.; et al. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J. Clin. Investig. 2005, 115, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetsugu, H.; Iimuro, Y.; Uehara, T.; Nishio, T.; Harada, N.; Yoshida, M.; Hatano, E.; Son, G.; Fujimoto, J.; Yamaoka, Y. Nuclear factor κB inactivation in the rat liver ameliorates short term total warm ischaemia/reperfusion injury. Gut 2005, 54, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Bradham, C.A.; Schemmer, P.; Stachlewitz, R.F.; Thurman, R.G.; Brenner, D.A. Activation of nuclear factor-κB during orthotopic liver transplantation in rats is protective and does not require Kupffer cells. Liver Transplant. Surg. 1999, 5, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Íimuro, Y.; Nishiura, T.; Hellerbrand, C.; Behrns, K.E.; Schoonhoven, R.; Grisham, J.W.; Brenner, D.A. NFκB prevents apoptosis and liver dysfunction during liver regeneration. J. Clin. Investig. 1998, 101, 802–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, H.; Lemasters, J.J. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology 2003, 125, 1246–1257. [Google Scholar] [CrossRef]
- Ben-Ari, Z.; Pappo, O.; Cheporko, Y.; Yasovich, N.; Offen, D.; Shainberg, A.; Leshem, D.; Sulkes, J.; Vidne, B.; Hochhauser, E. Bax Ablation Protects Against Hepatic Ischemia/Reperfusion Injury in Transgenic Mice. Liver Transplant. 2007, 13, 1181–1188. [Google Scholar] [CrossRef]
- Sabet Sarvestani, F.; Azarpira, N.; Al-Abdullah, I.H.; Tamaddon, A.M. microRNAs in liver and kidney ischemia reperfusion injury: Insight to improve transplantation outcome. Biomed. Pharmacother. 2021, 133, 110944. [Google Scholar] [CrossRef]
- Yu, C.H.; Xu, C.F.; Li, Y.M. Association of microRNA-223 expression with hepatic ischemia/reperfusion injury in mice. Dig. Dis. Sci. 2009, 54, 2362–2366. [Google Scholar] [CrossRef]
- Pan, W.; Wang, L.; Zhang, X.F.; Zhang, H.; Zhang, J.; Wang, G.; Xu, P.; Zhang, Y.; Hu, P.; Zhang, X.D.; et al. Hypoxia-induced microRNA-191 contributes to hepatic ischemia/reperfusion injury through the ZONAB/Cyclin D1 axis. Cell Death Differ. 2019, 26, 291–305. [Google Scholar] [CrossRef]
- Hao, W.; Zhao, Z.H.; Meng, Q.T.; Tie, M.E.; Lei, S.Q.; Xia, Z.Y. Propofol protects against hepatic ischemia/reperfusion injury via miR-133a-5p regulating the expression of MAPK6. Cell Biol. Int. 2017, 41, 495–504. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Wang, Z.; Wang, T.; Yu, Y.; He, J.; Zhang, H.; Yang, T.; Shen, Z. MicroRNA-17 regulates autophagy to promote hepatic ischemia/reperfusion injury via suppression of signal transductions and activation of transcription-3 expression. Liver Transplant. 2016, 22, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Mou, T.; Luo, Y.; Pu, X.; Pu, J.; Wan, L.; Gong, J.; Yang, H.; Liu, Y.; Li, Z.; et al. Inhibition of miR-450b-5p ameliorates hepatic ischemia/reperfusion injury via targeting CRYAB. Cell Death Dis. 2020, 11, 455. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, M.; Xu, L.N.; Yin, L.H.; Qi, Y.; Peng, J.Y. MicroRNA-142-3p attenuates hepatic ischemia/reperfusion injury via targeting of myristoylated alanine-rich C-kinase substrate. Pharmacol. Res. 2020, 156, 104783. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kong, L.; Xu, X.; Geng, Q.; Tang, W.; Jiang, W. Down-regulation of MicroRNA-146a in the early stage of liver ischemia-reperfusion injury. Transplant. Proc. 2013, 45, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Kong, L.; Ni, Q.; Lu, Y.; Ding, W.; Liu, G.; Pu, L.; Tang, W.; Kong, L. miR-146a ameliorates liver ischemia/reperfusion injury by suppressing IRAK1 and TRAF6. PLoS ONE 2014, 9, e101530. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gu, C.; Huang, X. Sevoflurane protects against hepatic ischemia/reperfusion injury by modulating microRNA-200c regulation in mice. Biomed. Pharmacother. 2016, 84, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Gao, Y.; Qin, J.; Lu, S. The role of miR-34a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS ONE 2014, 9, e113305. [Google Scholar] [CrossRef]
- Li, L.; Li, G.; Yu, C.; Shen, Z.; Xu, C.; Feng, Z.; Zhang, X.; Li, Y. A role of microRNA-370 in hepatic ischaemia-reperfusion injury by targeting transforming growth factor-β receptor II. Liver Int. 2015, 35, 1124–1132. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, F.; Song, W.; Zhang, B.; Zhang, X.; Jin, X.; Li, H. Altered miR-370 expression in hepatic ischemia-reperfusion injury correlates with the level of nuclear kappa B (NF-κB) related factors. Gene 2017, 607, 23–30. [Google Scholar] [CrossRef]
- Jiang, W.; Liu, G.; Tang, W. MicroRNA-182-5p Ameliorates Liver Ischemia-Reperfusion Injury by Suppressing Toll-like Receptor 4. Transplant. Proc. 2016, 48, 2809–2814. [Google Scholar] [CrossRef]
- Huang, Z.; Zheng, D.; Pu, J.; Dai, J.; Zhang, Y.; Zhang, W.; Wu, Z. MicroRNA-125b protects liver from ischemia/reperfusion injury via inhibiting TRAF6 and NF-κB pathway. Biosci. Biotechnol. Biochem. 2019, 83, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Mou, T.; Luo, Y.; Huang, Z.; Zheng, D.; Pu, X.; Shen, A.; Pu, J.; Li, T.; Dai, J.; Chen, W.; et al. Inhibition of microRNA-128-3p alleviates liver ischaemia–reperfusion injury in mice through repressing the Rnd3/NF-κB axis. Innate Immun. 2020, 26, 528–536. [Google Scholar] [CrossRef]
- Zhai, Y.; Busuttil, R.; Kupiec-Weglinski, J. Liver Ischemia and Reperfusion Injury: New Insights Into Mechanisms of Innate—Adaptive Immune-Mediated Tissue Inflammation. Am. J. Transpl. 2011, 11, 1563–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, R.; Rossetti, M.; Naini, B.; Groysberg, V.; Kaldas, F.; Busuttil, R.; Chang, Y.; Gjertson, D.; Kupiec-Weglinski, J.; Reed, E. Pattern Recognition Receptor-Reactivity Screening of Liver Transplant Patients: Potential for Personalized and Precise Organ Matching to Reduce Risks of Ischemia-Reperfusion Injury. Ann. Surg. 2020, 271, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Kim, M.; Novitsky, E.; D’Agati, V.; Lee, H.T. Intestinal TLR9 deficiency exacerbates hepatic IR injury via altered intestinal inflammation and short-chain fatty acid synthesis. FASEB J. 2020, 34, 12083–12099. [Google Scholar] [CrossRef]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4-dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef]
- Mendes-Braz, M.; Elias-Miró, M.; Jiménez-Castro, M.B.; Casillas-Ramírez, A.; Ramalho, F.S.; Peralta, C. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J. Biomed. Biotechnol. 2012, 2012, 8657. [Google Scholar] [CrossRef] [Green Version]
- Hanschen, M.; Zahler, S.; Krombach, F.; Khandoga, A. Reciprocal activation between CD4+ T cells and Kupffer cells during hepatic ischemia-reperfusion. Transplantation 2008, 86, 710–718. [Google Scholar] [CrossRef]
- Nakamitsu, A.; Hiyama, E.; Imamura, Y.; Matsuura, Y.; Yokoyama, T. Kupffer cell function in ischemic and nonischemic livers after hepatic partial ischemia/reperfusion. Surg. Today 2001, 31, 140–148. [Google Scholar] [CrossRef]
- Ye, L.; He, S.; Mao, X.; Zhang, Y.; Cai, Y.; Li, S. Effect of Hepatic Macrophage Polarization and Apoptosis on Liver Ischemia and Reperfusion Injury During Liver Transplantation. Front. Immunol. 2020, 11, 1193. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Okaya, T.; Martignoni, A.; Husted, T.; Schuster, R.; Lentsch, A.B. Divergent functions of CD4+ T lymphocytes in acute liver inflammation and injury after ischemia-reperfusion. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 289, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, G.; Rao, J.; Pu, L.; Yu, Y.; Wang, X.; Zhang, F. In vitro induced CD4+CD25+Foxp3+ Tregs attenuate hepatic ischemia-reperfusion injury. Int. Immunopharmacol. 2009, 9, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Zhang, R.; Gu, J.; Ju, Z.; Wu, X.; Wang, Q.; Peng, H.; Qiu, J.; Zhou, J.; Cheng, F.; et al. Acidic Microenvironment Regulates the Severity of Hepatic Ischemia/Reperfusion Injury by Modulating the Generation and Function of Tregs via the PI3K-mTOR Pathway. Front. Immunol. 2020, 10, 2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell-Kenkel, J.C.; Currin, R.T.; Tanaka, Y.; Thurman, R.G.; Lemasters, J.J. Reperfusion injury to endothelial cells following cold ischemic storage of rat livers. Hepatology 1989, 10, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Caldwell-Kenkel, J.C.; Currin, R.T.; Tanaka, Y.; Thurman, R.G.; Lemasters, J.J. Kupffer cell activation and endothelial cell damage after storage of rat livers: Effects of reperfusion. Hepatology 1991, 13, 83–95. [Google Scholar] [CrossRef]
- Gao, W.; Bentley, R.C.; Madden, J.E.; Clavien, P.A. Apoptosis of sinusoidal endothelial cells is a critical mechanism of preservation injury in rat liver transplantation. Hepatology 1998, 27, 1652–1660. [Google Scholar] [CrossRef]
- Jaeschke, H. Mechanisms of reperfusion injury after warm ischemia of the liver. J. Hepatobiliary Pancreat. Surg. 1998, 5, 402–408. [Google Scholar] [CrossRef]
- Teoh, N.; Farrell, G. Hepatic ischemia reperfusion injury: Pathogenic mechanisms and basis for hepatoprotection. J. Gastroenterol. Hepatol. 2003, 18, 891–902. [Google Scholar] [CrossRef]
- Teoh, N.C. Hepatic ischemia reperfusion injury: Contemporary perspectives on pathogenic mechanisms and basis for hepatoprotection-the good, bad and deadly. J. Gastroenterol. Hepatol. 2011, 26, 180–187. [Google Scholar] [CrossRef]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. J. Hepatol. 2012, 57, 692–694. [Google Scholar] [CrossRef]
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.I.; Mars, W.M.; Michalopoulos, G.K. Signals and cells involved in regulating liver regeneration. Cells 2012, 1, 1261–1292. [Google Scholar] [CrossRef] [Green Version]
- Kuboki, S.; Shin, T.; Huber, N.; Eismann, T.; Galloway, E.; Schuster, R.; Blanchard, J.; Edwards, M.; Lentsch, A. Hepatocyte Signaling through CXCR2 is Detrimental to Liver Recovery after Ischemia/Reperfusion in Mice. Hepatology 2008, 48, 1213–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, S.; Okaya, T.; Rudich, S.; Petrovic, S.; Tenrani, K.; Wang, Z.; Zahedi, K.; Casero, R.A.; Lentsch, A.B.; Soleimani, M. Distinct and sequential upregulation of genes regulating cell growth and cell cycle progression during hepatic ischemia-reperfusion injury. Am. J. Physiol.-Cell Physiol. 2005, 289, 826–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.C.; Kuboki, S.; Freeman, C.M.; Nojima, H.; Schuster, R.M.; Edwards, M.J.; Lentsch, A.B. CXC Chemokines Function as a Rheostat for Hepatocyte Proliferation and Liver Regeneration. PLoS ONE 2015, 10, e0120092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, K.; Horiuchi, T.; Uchinami, M.; Tabo, T.; Kimura, N.; Yokomachi, J.; Yoshida, M.; Tanaka, K. Hepatic ischemia-reperfusion promotes liver metastasis of colon cancer. J. Surg. Res. 2002, 105, 243–247. [Google Scholar] [CrossRef]
- Van Der Bilt, J.D.W.; Kranenburg, O.; Nijkamp, M.W.; Smakman, N.; Veenendaal, L.M.; Te Velde, E.A.; Voest, E.E.; Van Diest, P.J.; Borel Rinkes, I.H.M. Ischemia/reperfusion accelerates the outgrowth of hepatic micrometastases in a highly standardized murine model. Hepatology 2005, 42, 165–175. [Google Scholar] [CrossRef]
- Yamashita, S.; Venkatesan, A.M.; Mizuno, T.; Aloia, T.A.; Chun, Y.S.; Lee, J.E.; Vauthey, J.N.; Conrad, C. Remnant liver ischemia as a prognostic factor for cancer-specific Survival after resection of colorectal liver metastases. JAMA Surg. 2017, 152, e172986. [Google Scholar] [CrossRef]
- Nagai, S.; Yoshida, A.; Facciuto, M.; Moonka, D.; Abouljoud, M.S.; Schwartz, M.E.; Florman, S.S. Ischemia time impacts recurrence of hepatocellular carcinoma after liver transplantation. Hepatology 2015, 61, 895–904. [Google Scholar] [CrossRef]
- Nicoud, I.B.; Jones, C.M.; Pierce, J.M.; Earl, T.M.; Matrisian, L.M.; Chari, R.S.; Gorden, D.L. Warm hepatic ischemia-reperfusion promotes growth of colorectal carcinoma micrometastases in mouse liver via matrix metalloproteinase-9 induction. Cancer Res. 2007, 67, 2720–2728. [Google Scholar] [CrossRef]
- Wassmer, C.H.; Moeckli, B.; Berney, T.; Toso, C.; Orci, L.A. Shorter survival after liver pedicle clamping in patients undergoing liver resection for hepatocellular carcinoma revealed by a systematic review and meta-analysis. Cancers 2021, 13, 637. [Google Scholar] [CrossRef]
- Van Der Bilt, J.D.W.; Soeters, M.E.; Duyverman, A.M.M.J.; Nijkamp, M.W.; Witteveen, P.O.; Van Diest, P.J.; Kranenburg, O.; Rinkes, I.H.M.B. Perinecrotic hypoxia contributes to ischemia/reperfusion-accelerated outgrowth of colorectal micrometastases. Am. J. Pathol. 2007, 170, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Konishi, T.; Schuster, R.M.; Goetzman, H.S.; Caldwell, C.C.; Lentsch, A.B. Fibrotic liver has prompt recovery after ischemia-reperfusion injury. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, G390–G400. [Google Scholar] [CrossRef] [PubMed]
- Konishi, T.; Schuster, R.M.; Lentsch, A.B. Liver repair and regeneration after ischemia-reperfusion injury is associated with prolonged fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2019, 316, G323–G331. [Google Scholar] [CrossRef]
- Cursio, R.; Gugenheim, J. Ischemia-Reperfusion Injury and Ischemic-Type Biliary Lesions following Liver Transplantation. J. Transplant. 2012, 2012, 164329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Pang, L.; Guo, Y.; Ke, N.; Li, S.; Wei, L.; Li, Q.; Li, Y. Hypoxia/reoxygenation up-regulates death receptor expression and enhances apoptosis in human biliary epithelial cells. Life Sci. 2009, 85, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Vierling, J.M.; Fennell, R.H. Histopathology of early and late human hepatic allograft rejection: Evidence of progressive destruction of interlobular bile ducts. Hepatology 1985, 5, 1076–1082. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: A 2015 update. Clin. Sci. 2015, 129, 345–362. [Google Scholar] [CrossRef]
- Gertsch, P.; Vandoni, R.E.; Pelloni, A.; Krpo, A.; Alerci, M. Localized hepatic ischemia after liver resection: A prospective evaluation. Ann. Surg. 2007, 246, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, L.T.; Van Trigt, J.D.; Reiniers, M.J.; Busch, O.R.; Gouma, D.J.; Van Gulik, T.M. Vascular occlusion or not during liver resection: The continuing story. Dig. Surg. 2012, 29, 35–42. [Google Scholar] [CrossRef]
- Radermacher, P.; Billiar, T.R.; Ghezzi, P.; Martin, L.; Thiemermann, C. Editorial: Translational Insights Into Mechanisms and Therapy of Organ Dysfunction in Sepsis and Trauma. Front. Immunol. 2020, 11, 1987. [Google Scholar] [CrossRef] [PubMed]
- Messerer, D.A.C.; Gaessler, H.; Hoffmann, A.; Gröger, M.; Benz, K.; Huhn, A.; Hezel, F.; Calzia, E.; Radermacher, P.; Datzmann, T. The H2S Donor Sodium Thiosulfate (Na2S2O3) Does Not Improve Inflammation and Organ Damage After Hemorrhagic Shock in Cardiovascular Healthy Swine. Front. Immunol. 2022, 13, 901005. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Yamaguchi, Y.; Matsumura, F.; Akizuki, E.; Okabe, K.; Liang, J.; Ohshiro, H.; Ichiguchi, O.; Yamada, S.; Mori, K.; et al. Immunosuppressants Decrease Neutrophil Chemoattractant and Attenuate Ischemia/Reperfusion Injury of the Liver in Rats. Trauma Acute Care Surg. 1998, 44, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Askar, I.; Bozkurt, M. Protective effects of immunosuppressants and steroids against ischemia-reperfusion injury in cremaster muscle flap at microcirculatory level. Microsurgery 2002, 22, 361–366. [Google Scholar] [CrossRef]
- Arias-Diaz, J.; Ildefonso, J.A.; Mũoz, J.J.; Zapata, A.; Jiménez, E. Both tacrolimus and sirolimus decrease Th1Th2 ratio, and increase regulatory T lymphocytes in the liver after ischemia/reperfusion. Lab. Investig. 2009, 89, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Hirao, H.; Nakamura, K.; Kupiec-Weglinski, J.W. Liver ischaemia–reperfusion injury: A new understanding of the role of innate immunity. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 239–256. [Google Scholar] [CrossRef]
- Barzegar, M.; Kaur, G.; Gavins, F.N.E.; Wang, Y.; Boyer, C.J.; Alexander, J.S. Potential therapeutic roles of stem cells in ischemia-reperfusion injury. Stem Cell Res. 2019, 37, 101421. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, T.; Ren, J.; Xia, Y.; Onuma, A.; Wang, Y.; He, J.; Wu, J.; Wang, H.; Hamad, A.; et al. Pre-operative exercise therapy triggers anti-inflammatory trained immunity of Kupffer cells through metabolic reprogramming. Nat. Metab. 2021, 3, 843–858. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pretzsch, E.; Nieß, H.; Khaled, N.B.; Bösch, F.; Guba, M.; Werner, J.; Angele, M.; Chaudry, I.H. Molecular Mechanisms of Ischaemia-Reperfusion Injury and Regeneration in the Liver-Shock and Surgery-Associated Changes. Int. J. Mol. Sci. 2022, 23, 12942. https://doi.org/10.3390/ijms232112942
Pretzsch E, Nieß H, Khaled NB, Bösch F, Guba M, Werner J, Angele M, Chaudry IH. Molecular Mechanisms of Ischaemia-Reperfusion Injury and Regeneration in the Liver-Shock and Surgery-Associated Changes. International Journal of Molecular Sciences. 2022; 23(21):12942. https://doi.org/10.3390/ijms232112942
Chicago/Turabian StylePretzsch, Elise, Hanno Nieß, Najib Ben Khaled, Florian Bösch, Markus Guba, Jens Werner, Martin Angele, and Irshad H. Chaudry. 2022. "Molecular Mechanisms of Ischaemia-Reperfusion Injury and Regeneration in the Liver-Shock and Surgery-Associated Changes" International Journal of Molecular Sciences 23, no. 21: 12942. https://doi.org/10.3390/ijms232112942