
Unraveling Mitochondrial Determinants of Tumor Response to Radiation Therapy
 , , ,
, , , 
Abstract
:1. Introduction
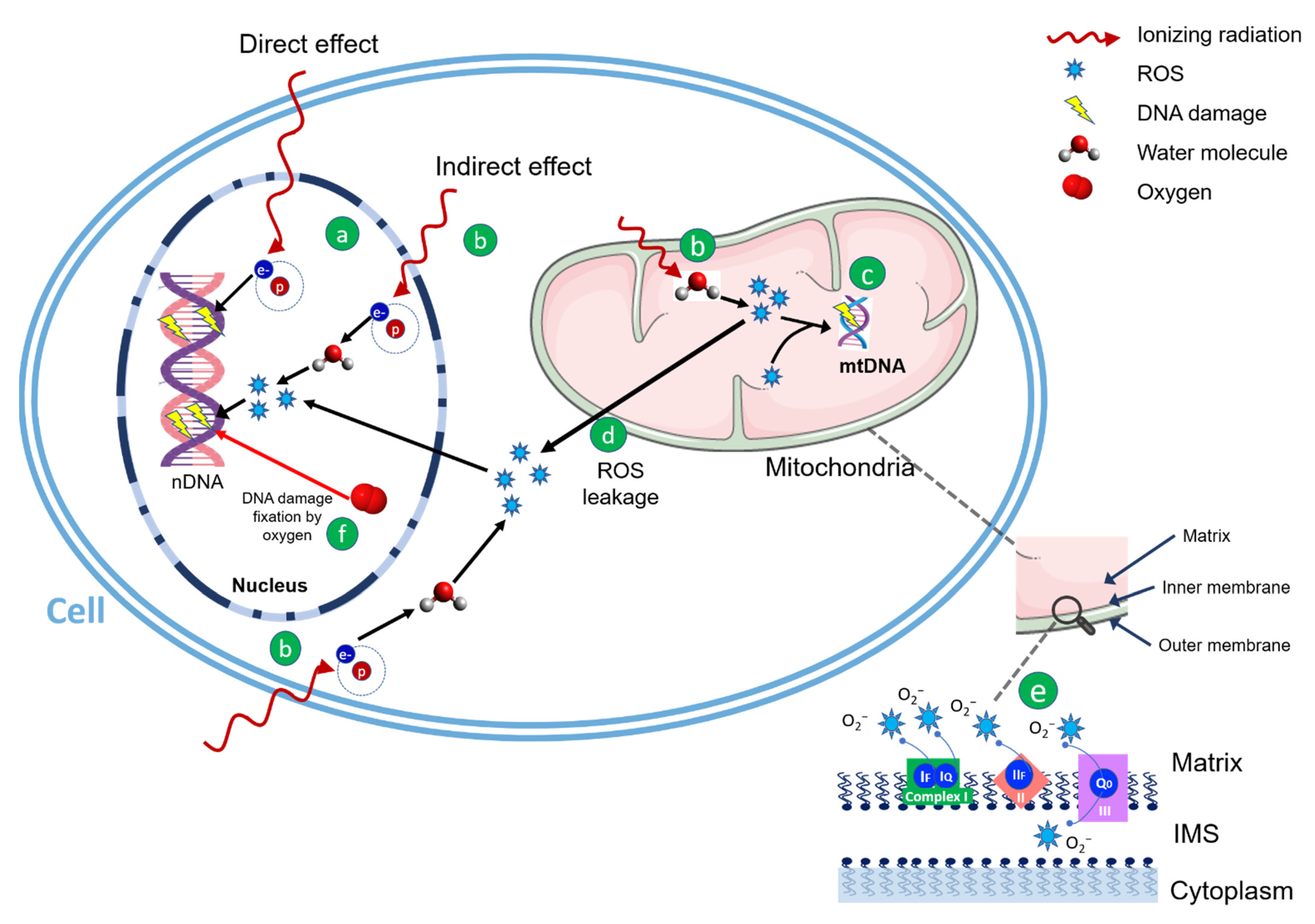
2. Radiation Therapy and Mitochondria
3. Effect of Radiation Quality on ROS Generation
4. Radioresistance and Mitochondria: Roles of Hypoxia and Metabolic Alterations
5. Mitochondria and Epigenetics
6. Mitochondria-Targeting Strategies to Improve RT Effects

{kind=link}
| Compound Name | Mitochondria-Targeted Unit | Testing Models | References |
|---|---|---|---|
| Lonidamine | complex II | human glioma cell lines | Prabhakara et al. [92], 2018 |
| human glioma cell lines | Kalia, V et al. [93], 2009 | ||
| non-small cell lung cancer cell lines | Meijer T W H et al. [94], 2018 | ||
| Non-steroidal anti-inflammatory drugs | complex I | murine TLT liver tumors and FSaII fibrosarcomas | Crokart et al. [98], 2005 |
| Glucocorticoids | complex I and complex III | murine TLT liver tumors and FSaII fibrosarcomas | Crokart et al. [99], 2007 |
| Metformin | complex I | prostate cancer | Zannella VE et al. [96], 2013 |
| prostate cancer | Taira AV et al. [112], 2014 | ||
| rectal cancer | Skinner HD et al. [113], 2013 | ||
| esophageal cancer | Spierings et al. [114], 2015; | ||
| liver cancer | Jang et al. [115], 2015 | ||
| head and neck cancer | Spratt et al. [116], 2016 | ||
| Atovaquone | complex III | FaDU and HCT116 xenografts in nude mice | Ashton et al. [102], 2016 |
| non-small cell lung cancer | ARCADIAN TRIAL (currently in recruiting phase) | ||
| Auranofin | mitochondrial thioredoxin reductase | H1299 tumor cell lines xenografted in murine models | Wang et al. [103], 2017 |
7. Conclusions and Future Perspectives
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Hayashi, J.-I. A novel function of mtDNA: Its involvement in metastasis. Ann. N. Y. Acad. Sci. 2010, 1201, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Turnbull, D.M.; Minczuk, M.; Gammage, P.A. Therapeutic Manipulation of mtDNA Heteroplasmy: A Shifting Perspective. Trends Mol. Med. 2020, 26, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. Roles of mitochondria in the hallmarks of metastasis. Br. J. Cancer 2021, 124, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Izzo, V.; Bravo San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Lynam-Lennon, N.; Maher, S.; Maguire, A.; Phelan, J.; Muldoon, C.; Reynolds, J.V.; O’Sullivan, J. Altered Mitochondrial Function and Energy Metabolism Is Associated with a Radioresistant Phenotype in Oesophageal Adenocarcinoma. PLoS ONE 2014, 9, e100738. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 87. [Google Scholar] [CrossRef] [PubMed]
- McCann, E.; O’Sullivan, J.; Marcone, S. Targeting cancer-cell mitochondria and metabolism to improve radiotherapy response. Transl. Oncol. 2021, 14, 100905. [Google Scholar] [CrossRef] [PubMed]
- Jameel, J.; Rao, V.S.; Cawkwell, L.; Drew, P. Radioresistance in carcinoma of the breast. Breast 2004, 13, 452–460. [Google Scholar] [CrossRef]
- Ahmed, K.A.; Chinnaiyan, P.; Fulp, W.J.; Eschrich, S.; Torres-Roca, J.F.; Caudell, J.J. The radiosensitivity index predicts for overall survival in glioblastoma. Oncotarget 2015, 6, 34414–34422. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Morgan, W.F. Non-targeted and Delayed Effects of Exposure to Ionizing Radiation: I. Radiation-Induced Genomic Instability and Bystander Effects In Vitro. Radiat. Res. 2003, 159, 567–580. [Google Scholar] [CrossRef]
- Morgan, W.F. Non-targeted and Delayed Effects of Exposure to Ionizing Radiation: II. Radiation-Induced Genomic Instability and Bystander Effects In Vivo, Clastogenic Factors and Transgenerational Effects. Radiat. Res. 2003, 159, 581–596. [Google Scholar] [CrossRef]
- Spitz, D.R.; Hauer-Jensen, M. Ionizing Radiation-Induced Responses: Where Free Radical Chemistry Meets Redox Biology and Medicine. Antioxidants Redox Signal. 2014, 20, 1407–1409. [Google Scholar] [CrossRef]
- Clutton, S.; Townsend, K.; Walker, C.; Ansell, J.; Wright, E. Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis 1996, 17, 1633–1639. [Google Scholar] [CrossRef]
- Leach, J.K.; Van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar] [PubMed]
- Szumiel, I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: The pivotal role of mitochondria. Int. J. Radiat. Biol. 2015, 91, 934929. [Google Scholar] [CrossRef] [PubMed]
- Chatzipapas, K.P.; Papadimitroulas, P.; Emfietzoglou, D.; Kalospyros, S.A.; Hada, M.; Georgakilas, A.G.; Kagadis, G.C. Ionizing Radiation and Complex DNA Damage: Quantifying the Radiobiological Damage Using Monte Carlo Simulations. Cancers 2020, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Ewing, D. The oxygen fixation hypothesis: A reevaluation. Am. J. Clin. Oncol. 1998, 21, 355–361. [Google Scholar] [CrossRef]
- Tharmalingham, H.; Hoskin, P. Clinical trials targeting hypoxia. Br. J. Radiol. 2019, 92, 20170966. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Azzam, E.I.; Li, J.J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Averbeck, D.; Rodriguez-Lafrasse, C. Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. Int. J. Mol. Sci. 2021, 22, 11047. [Google Scholar] [CrossRef]
- Zhou, Z.; Song, J.; Nie, L.; Chen, X. Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem. Soc. Rev. 2016, 45, 6597–6626. [Google Scholar] [CrossRef]
- Gao, L.; Laude, K.; Cai, H. Mitochondrial Pathophysiology, Reactive Oxygen Species, and Cardiovascular Diseases. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 137–155. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, H.; De Ridder, M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett. 2018, 438, 154–164. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2014, 356, 156–164. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Giampazolias, E.; Tait, S.W. Mitochondria and the hallmarks of cancer. FEBS J. 2016, 283, 803–814. [Google Scholar] [CrossRef]
- Richardson, R.B.; Harper, M.-E. Mitochondrial stress controls the radiosensitivity of the oxygen effect: Implications for radiotherapy. Oncotarget 2016, 7, 21469–21483. [Google Scholar] [CrossRef]
- Guo, X.; Yang, N.; Ji, W.; Zhang, H.; Dong, X.; Zhou, Z.; Li, L.; Shen, H.; Yao, S.Q.; Huang, W. Mito-Bomb: Targeting Mitochondria for Cancer Therapy. Adv. Mater. 2021, 33, 2007778. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Hada, M.; Wu, H.; Cucinotta, F.A. mBAND analysis for high- and low-LET radiation-induced chromosome aberrations: A review. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Paganetti, H. Relative biological effectiveness (RBE) values for proton beam therapy. Variations as a function of biological endpoint, dose, and linear energy transfer. Phys. Med. Biol. 2014, 59, R419–R472. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; LeDuc, A.; Pottier, I.; Prevost, V.; Sichel, F.; Lefaix, J.-L. Dramatic Increase in Oxidative Stress in Carbon-Irradiated Normal Human Skin Fibroblasts. PLoS ONE 2013, 8, e85158. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Breda, C.N.D.S.; Davanzo, G.G.; Basso, P.J.; Câmara, N.O.S.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Galluzzi, L. Mitochondrial control of innate immune signaling by irradiated cancer cells. OncoImmunology 2020, 9, 1797292. [Google Scholar] [CrossRef]
- Durante, M.; Orecchia, R.; Loeffler, J.S. Charged-particle therapy in cancer: Clinical uses and future perspectives. Nat. Rev. Clin. Oncol. 2017, 14, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Gao, J.; Hu, J.; Lu, R.; Yang, J.; Qiu, X.; Hu, W.; Lu, J.J. Carbon ion radiotherapy boost in the treatment of glioblastoma: A randomized phase I/III clinical trial. Cancer Commun. 2019, 39, 5. [Google Scholar] [CrossRef]
- Keisari, Y.; Kelson, I. The Potentiation of Anti-Tumor Immunity by Tumor Abolition with Alpha Particles, Protons, or Carbon Ion Radiation and Its Enforcement by Combination with Immunoadjuvants or Inhibitors of Immune Suppressor Cells and Checkpoint Molecules. Cells 2021, 10, 228. [Google Scholar] [CrossRef]
- Marcus, D.; Lieverse, R.; Klein, C.; Abdollahi, A.; Lambin, P.; Dubois, L.; Yaromina, A. Charged Particle and Conventional Radiotherapy: Current Implications as Partner for Immunotherapy. Cancers 2021, 13, 1468. [Google Scholar] [CrossRef]
- Sato, H.; Demaria, S.; Ohno, T. The role of radiotherapy in the age of immunotherapy. Jpn. J. Clin. Oncol. 2021, 51, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Szumiel, I. Intrinsic Radiation Sensitivity: Cellular Signaling is the Key. Radiat. Res. 2008, 169, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Pitroda, S.P.; Wakim, B.T.; Sood, R.F.; Beveridge, M.G.; Beckett, M.A.; MacDermed, D.M.; Weichselbaum, R.R.; Khodarev, N.N. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. 2009, 7, 68. [Google Scholar] [CrossRef]
- Shimura, T.; Noma, N.; Sano, Y.; Ochiai, Y.; Oikawa, T.; Fukumoto, M.; Kunugita, N. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother. Oncol. 2014, 112, 302–307. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Sun, Y.; Li, J.; Wang, Y.; Zhu, Y.; Shi, Y.; Fan, X.; Zhou, J.; Bao, Y.; Xiao, J.; et al. Silencing the Girdin gene enhances radio-sensitivity of hepatocellular carcinoma via suppression of glycolytic metabolism. J. Exp. Clin. Cancer Res. 2017, 36, 110. [Google Scholar] [CrossRef]
- Semenza, G.; Roth, P.; Fang, H.; Wang, G. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Dachs, G.U.; Gleadle, J.M.; Nicholls, L.G.; Harris, A.L.; Stratford, I.J.; Hankinson, O.; Pugh, C.W.; Ratcliffe, P.J. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1997, 94, 8104–8109. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Briston, T.; Yang, J.; Ashcroft, M. HIF-1α localization with mitochondria: A new role for an old favorite? Cell Cycle Georget. Tex. 2011, 10, 4170–4171. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Heiden, M.G.V.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Mudassar, F.; Shen, H.; O’Neill, G.; Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 208. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Nejad, A.E.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Javanmard, S.H.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Hirayama, R. Mechanism of oxygen effect for photon and heavy-ion beams. Igaku Butsuri Nihon Igaku Butsuri Gakkai kikanshi Jpn. J. Med. Phys. Off. J. Jpn. Soc. Med. Phys. 2014, 34, 65–69. [Google Scholar]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. Berl. Ger. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, R.; Depping, R.; Delaperriere, M.; Dunst, J. Targeting hypoxia to overcome radiation resistance in head & neck cancers: Real challenge or clinical fairytale? Expert Rev. Anticancer Ther. 2016, 16, 751–758. [Google Scholar] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Bol, V.; Bol, A.; Bouzin, C.; Labar, D.; Lee, J.A.; Janssens, G.; Porporato, P.E.; Sonveaux, P.; Feron, O.; Grégoire, V. Reprogramming of tumor metabolism by targeting mitochondria improves tumor response to irradiation. Acta Oncol. 2015, 54, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Wu, C.T.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, Mechanisms and Clinical Perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Peschansky, V.; Wahlestedt, C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 2014, 9, 3–12. [Google Scholar] [CrossRef]
- Belli, M.; Indovina, L. The Response of Living Organisms to Low Radiation Environment and Its Implications in Radiation Protection. Front. Public Health 2020, 8, 601711. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-Da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, Â. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef] [PubMed]
- Mechta, M.; Ingerslev, L.R.; Fabre, O.; Picard, M.; Barrès, R. Evidence Suggesting Absence of Mitochondrial DNA Methylation. Front. Genet. 2017, 8, 166. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Kam, W.W.-Y.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Miousse, I.R.; Tobacyk, J.; Melnyk, S.; James, S.J.; Cheema, A.K.; Boerma, M.; Hauer-Jensen, M.; Koturbash, I. One-carbon metabolism and ionizing radiation: A multifaceted interaction. Biomol. Concepts 2017, 8, 83–92. [Google Scholar] [CrossRef]
- Baulch, J.E. Radiation-induced genomic instability, epigenetic mechanisms and the mitochondria: A dysfunctional ménage a trois? Int. J. Radiat. Biol. 2019, 95, 516–525. [Google Scholar] [CrossRef]
- Miousse, I.R.; Chang, J.; Shao, L.; Pathak, R.; Nzabarushimana, É.; Kutanzi, K.R.; Landes, R.D.; Tackett, A.J.; Hauer-Jensen, M.; Zhou, D.; et al. Inter-Strain Differences in LINE-1 DNA Methylation in the Mouse Hematopoietic System in Response to Exposure to Ionizing Radiation. Int. J. Mol. Sci. 2017, 18, 1430. [Google Scholar] [CrossRef]
- Guo, L.; Shestov, A.A.; Worth, A.J.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Glickson, J.D.; Blair, I.A. Inhibition of Mitochondrial Complex II by the Anticancer Agent Lonidamine. J. Biol. Chem. 2016, 291, 42–57. [Google Scholar] [CrossRef]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta 2016, 1866, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakara, S.; Kalia, V.K. Optimizing radiotherapy of brain tumours by a combination of temozolomide & lonidamine. Indian J. Med. Res. 2008, 128, 140. [Google Scholar] [PubMed]
- Kalia, V.; Prabhakara, S.; Narayanan, V. Modulation of cellular radiation responses by 2-deoxy-D-glucose and other glycolytic inhibitors: Implications for cancer therapy. J. Cancer Res. Ther. 2009, 5, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.W.; Peeters, W.J.; Dubois, L.J.; van Gisbergen, M.W.; Biemans, R.; Venhuizen, J.-H.; Span, P.N.; Bussink, J. Targeting glucose and glutamine metabolism combined with radiation therapy in non-small cell lung cancer. Lung Cancer 2018, 126, 32–40. [Google Scholar] [CrossRef]
- Miyato, Y.; Ando, K. Apoptosis of Human Melanoma Cells by a Combination of Lonidamine and Radiation. J. Radiat. Res. 2004, 45, 189–194. [Google Scholar] [CrossRef]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M.; et al. Reprogramming Metabolism with Metformin Improves Tumor Oxygenation and Radiotherapy Response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef]
- Lin, A.; Maity, A. Molecular Pathways: A Novel Approach to Targeting Hypoxia and Improving Radiotherapy Efficacy via Reduction in Oxygen Demand. Clin. Cancer Res. 2015, 21, 1995–2000. [Google Scholar] [CrossRef]
- Crokart, N.; Radermacher, K.; Jordan, B.F.; Baudelet, C.; Cron, G.O.; Grégoire, V.; Beghein, N.; Bouzin, C.; Feron, O.; Gallez, B. Tumor Radiosensitization by Antiinflammatory Drugs: Evidence for a New Mechanism Involving the Oxygen Effect. Cancer Res. 2005, 65, 7911–7916. [Google Scholar] [CrossRef]
- Crokart, N.; Jordan, B.F.; Baudelet, C.; Cron, G.O.; Hotton, J.; Radermacher, K.; Grégoire, V.; Beghein, N.; Martinive, P.; Bouzin, C.; et al. Glucocorticoids Modulate Tumor Radiation Response through a Decrease in Tumor Oxygen Consumption. Clin. Cancer Res. 2007, 13, 630–635. [Google Scholar] [CrossRef]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Rao, M.; Gao, C.; Guo, M.; Law, B.Y.K.; Xu, Y. Effects of metformin treatment on radiotherapy efficacy in patients with cancer and diabetes: A systematic review and meta-analysis. Cancer Manag. Res. 2018, 10, 4881–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef]
- Wang, H.; Bouzakoura, S.; de Mey, S.; Jiang, H.; Law, K.; Dufait, I.; Corbet, C.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Auranofin radiosensitizes tumor cells through targeting thioredoxin reductase and resulting overproduction of reactive oxygen species. Oncotarget 2017, 8, 35728–35742. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.F.; de Almeida, D.R.Q.; Terra, L.F.; Baptista, M.S.; Labriola, L. Photodynamic therapy in cancer treatment-an update review. J. Cancer Metastasis Treat. 2019, 5, 25. [Google Scholar] [CrossRef]
- Felsher, D.W. Cancer revoked: Oncogenes as therapeutic targets. Nat. Rev. Cancer 2003, 3, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Lei, Q.; Wang, S.-B.; Hu, J.-J.; Qiu, W.-X.; Zhu, J.-Y.; Yin, W.-N.; Luo, X.; Zhang, X.-Z. Dual-Stage-Light-Guided Tumor Inhibition by Mitochondria-Targeted Photodynamic Therapy. Adv. Funct. Mater. 2015, 25, 2961–2971. [Google Scholar] [CrossRef]
- Jaque, D.; Maestro, L.M.; del Rosal, B.; Haro-Gonzalez, P.; Benayas, A.; Plaza, J.L.; Rodríguez, E.M.; Solé, J.G. Nanoparticles for photothermal therapies. Nanoscale 2014, 6, 9494–9530. [Google Scholar] [CrossRef]
- Jung, H.S.; Han, J.; Lee, J.-H.; Lee, J.H.; Choi, J.-M.; Kweon, H.-S.; Han, J.H.; Kim, J.-H.; Byun, K.M.; Jung, J.H.; et al. Enhanced NIR Radiation-Triggered Hyperthermia by Mitochondrial Targeting. J. Am. Chem. Soc. 2015, 137, 3017–3023. [Google Scholar] [CrossRef]
- Jung, H.S.; Lee, J.-H.; Kim, K.; Koo, S.; Verwilst, P.; Sessler, J.L.; Kang, C.; Kim, J.S. A Mitochondria-Targeted Cryptocyanine-Based Photothermogenic Photosensitizer. J. Am. Chem. Soc. 2017, 139, 9972–9978. [Google Scholar] [CrossRef]
- Wang, G.D.; Nguyen, H.T.; Chen, H.; Cox, P.B.; Wang, L.; Nagata, K.; Hao, Z.; Wang, A.; Li, Z.; Xie, J. X-Ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy. Theranostics 2016, 6, 2295–2305. [Google Scholar] [CrossRef]
- Cho, W.; Kessel, D.; Rakowski, J.; Loughery, B.; Najy, A.; Pham, T.; Kim, S.; Kwon, Y.; Kato, I.; Kim, H.; et al. Photodynamic Therapy as a Potent Radiosensitizer in Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 1193. [Google Scholar] [CrossRef] [PubMed]
- Taira, A.V.; Merrick, G.S.; Galbreath, R.W.; Morris, M.; Butler, W.M.; Adamovich, E. Metformin is not associated with improved biochemical free survival or cause-specific survival in men with prostate cancer treated with permanent interstitial brachytherapy. J. Contemp. Brachytherapy 2014, 6, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Skinner, H.D.; McCurdy, M.R.; Echeverria, A.E.; Lin, S.H.; Welsh, J.W.; O’Reilly, M.S.; Hofstetter, W.L.; Ajani, J.A.; Komaki, R.; Cox, J.D.; et al. Metformin use and improved response to therapy in esophageal adenocarcinoma. Acta Oncol. 2013, 52, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Spierings, L.E.; Lagarde, S.M.; van Oijen, M.G.; Gisbertz, S.S.; Wilmink, J.W.; Hulshof, M.C.; Meijer, S.L.; Anderegg, M.C.; van Berge Henegouwen, M.I.; van Laarhoven, H.W. Metformin Use During Treatment of Potentially Curable Esophageal Cancer Patients is not Associated with Better Outcomes. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S766–S771. [Google Scholar] [CrossRef]
- Jang, W.I.; Kim, M.S.; Lim, J.S.; Yoo, H.J.; Seo, Y.S.; Han, C.J.; Han, C.J.; Park, S.C.; Kay, C.S.; Kim, M.; et al. Survival advantage associated with metformin usage in hepatocellular carcinoma patients receiving radiotherapy: A propensity score matching analysis. Anticancer Res. 2015, 35, 5047–5054. [Google Scholar]
- Spratt, D.E.; Zhang, C.; Zumsteg, Z.S.; Pei, X.; Zhang, Z.; Zelefsky, M.J. Metformin and prostate cancer: Reduced development of castration-resistant disease and prostate cancer mortality. Eur. Urol. 2013, 63, 709–716. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaffaroni, M.; Vincini, M.G.; Corrao, G.; Marvaso, G.; Pepa, M.; Viglietto, G.; Amodio, N.; Jereczek-Fossa, B.A. Unraveling Mitochondrial Determinants of Tumor Response to Radiation Therapy. Int. J. Mol. Sci. 2022, 23, 11343. https://doi.org/10.3390/ijms231911343
Zaffaroni M, Vincini MG, Corrao G, Marvaso G, Pepa M, Viglietto G, Amodio N, Jereczek-Fossa BA. Unraveling Mitochondrial Determinants of Tumor Response to Radiation Therapy. International Journal of Molecular Sciences. 2022; 23(19):11343. https://doi.org/10.3390/ijms231911343
Chicago/Turabian StyleZaffaroni, Mattia, Maria Giulia Vincini, Giulia Corrao, Giulia Marvaso, Matteo Pepa, Giuseppe Viglietto, Nicola Amodio, and Barbara Alicja Jereczek-Fossa. 2022. "Unraveling Mitochondrial Determinants of Tumor Response to Radiation Therapy" International Journal of Molecular Sciences 23, no. 19: 11343. https://doi.org/10.3390/ijms231911343