

The Proteoglycan Glypican-1 as a Possible Candidate for Innovative Targeted Therapeutic Strategies for Pancreatic Ductal Adenocarcinoma

Abstract
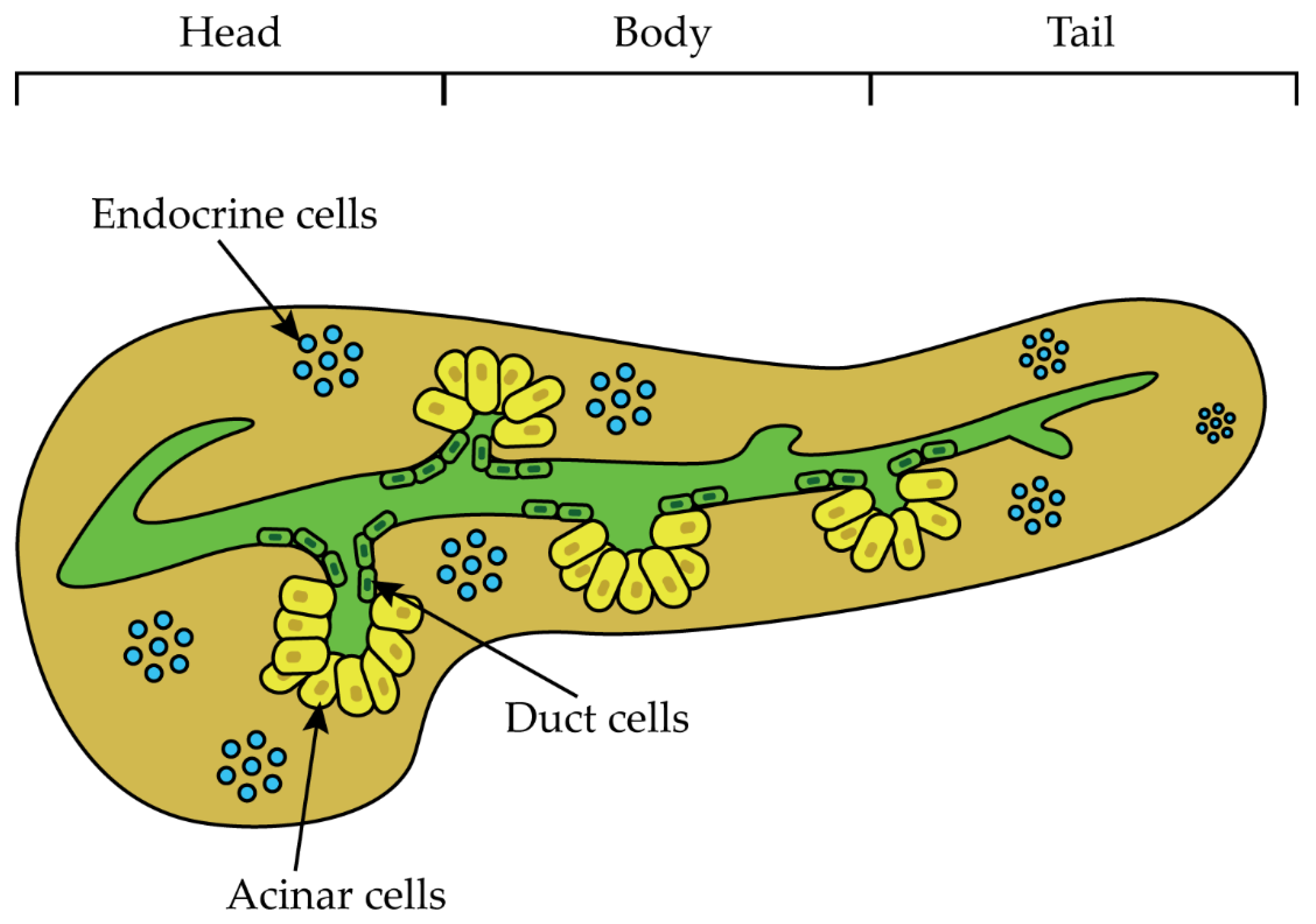
:1. Introduction
1.1. PDAC Risk Factors
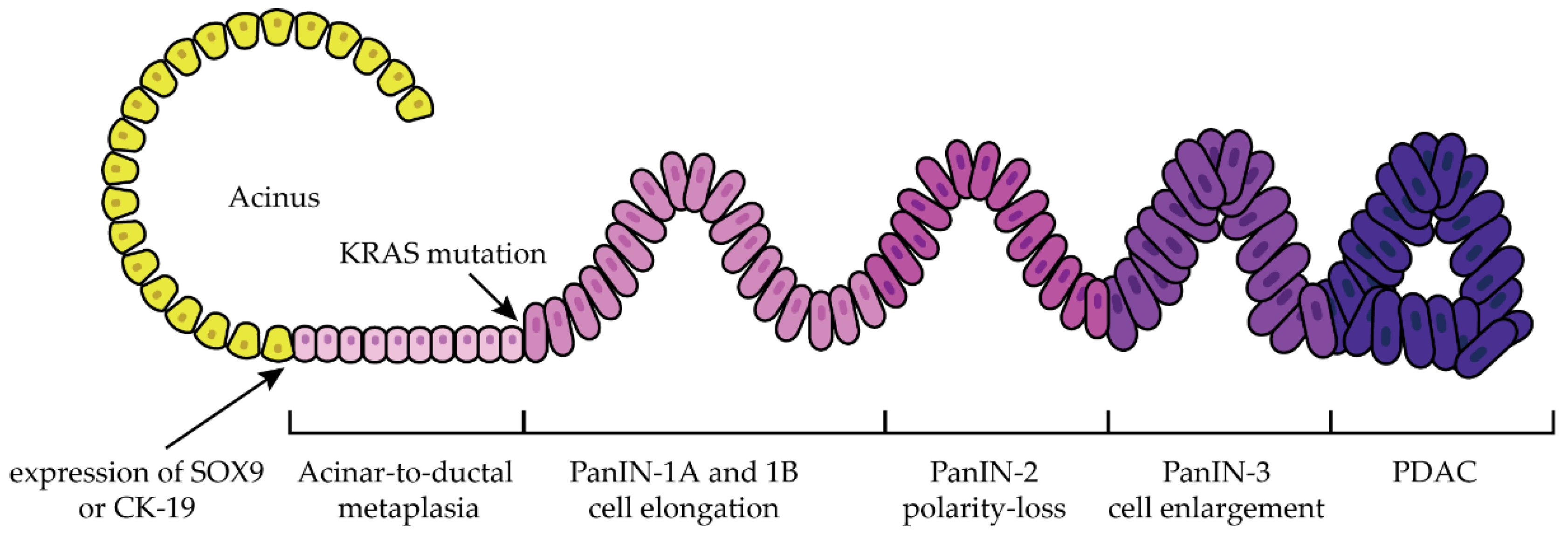
1.2. Progression
1.3. Diagnosis, Staging, and Treatment
2. Glypican-1 as Target Protein
2.1. Glypican Family
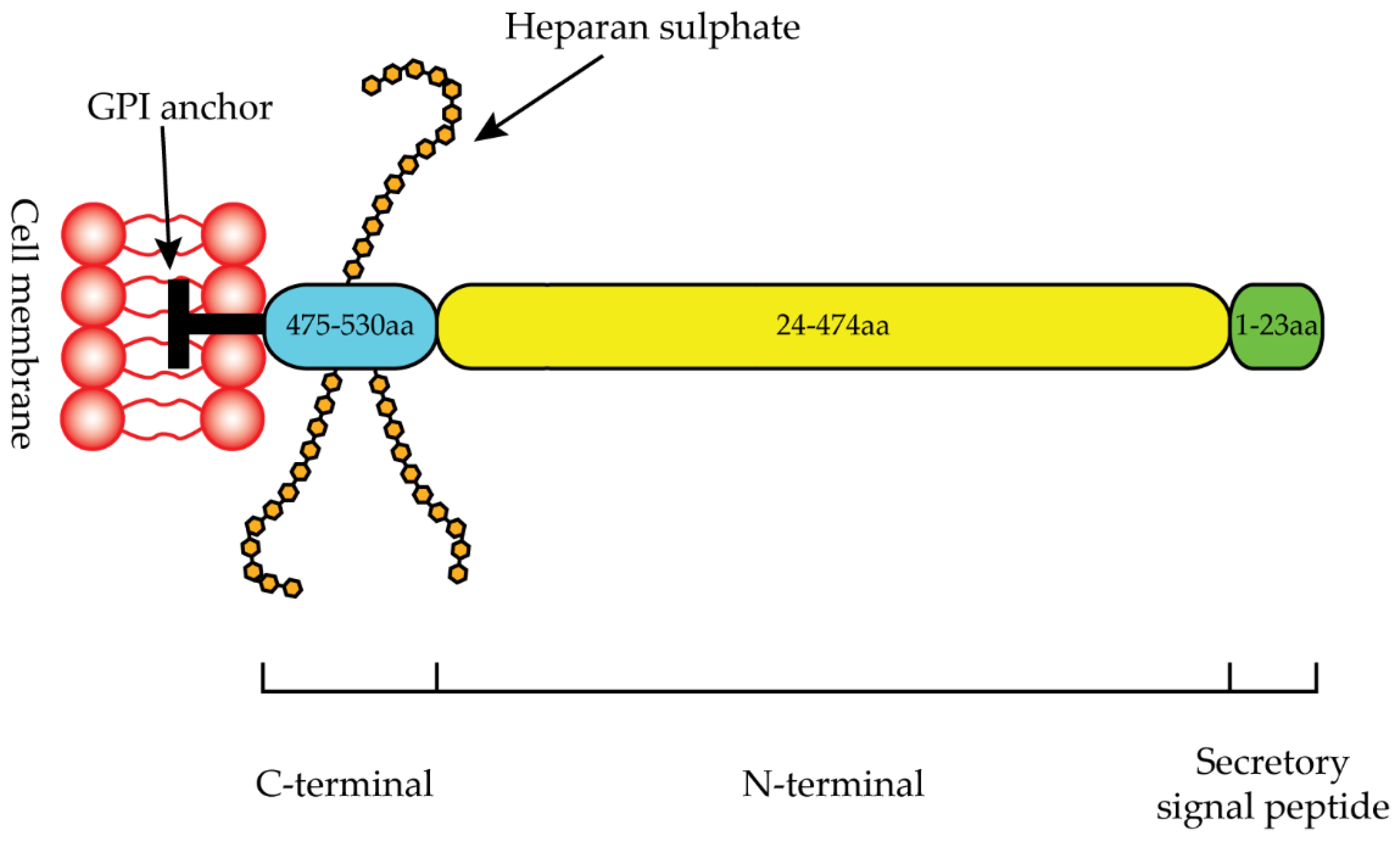
2.2. Glypican-1
2.3. Glypican-1 in PDAC
3. Targeting Strategies
3.1. Antibody-Based Immunotherapy
3.2. GPC1 as Target for Antibody-Based Immunotherapy in PDAC
3.3. CAR-T Cells
3.4. Antibody-Drug Conjugate
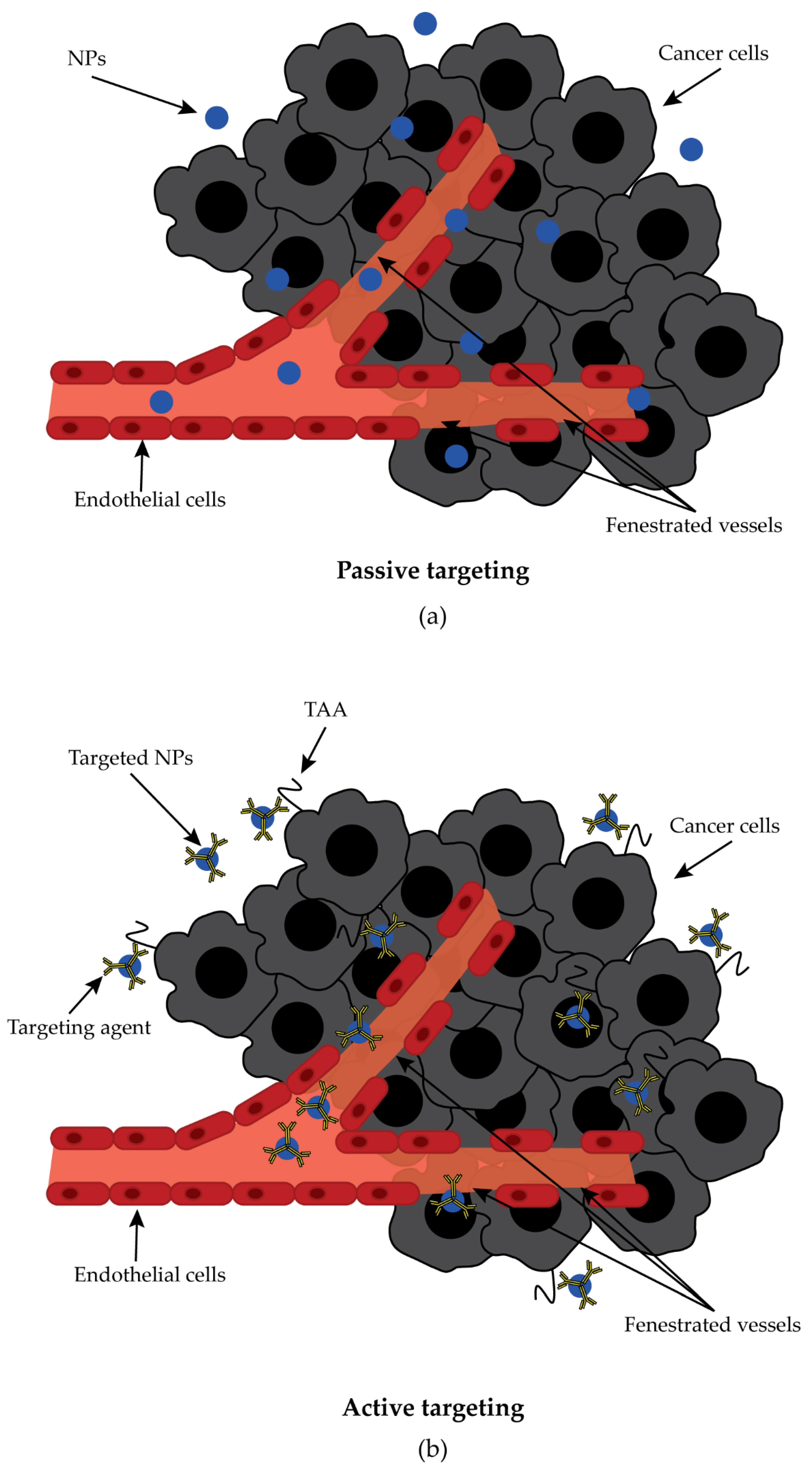
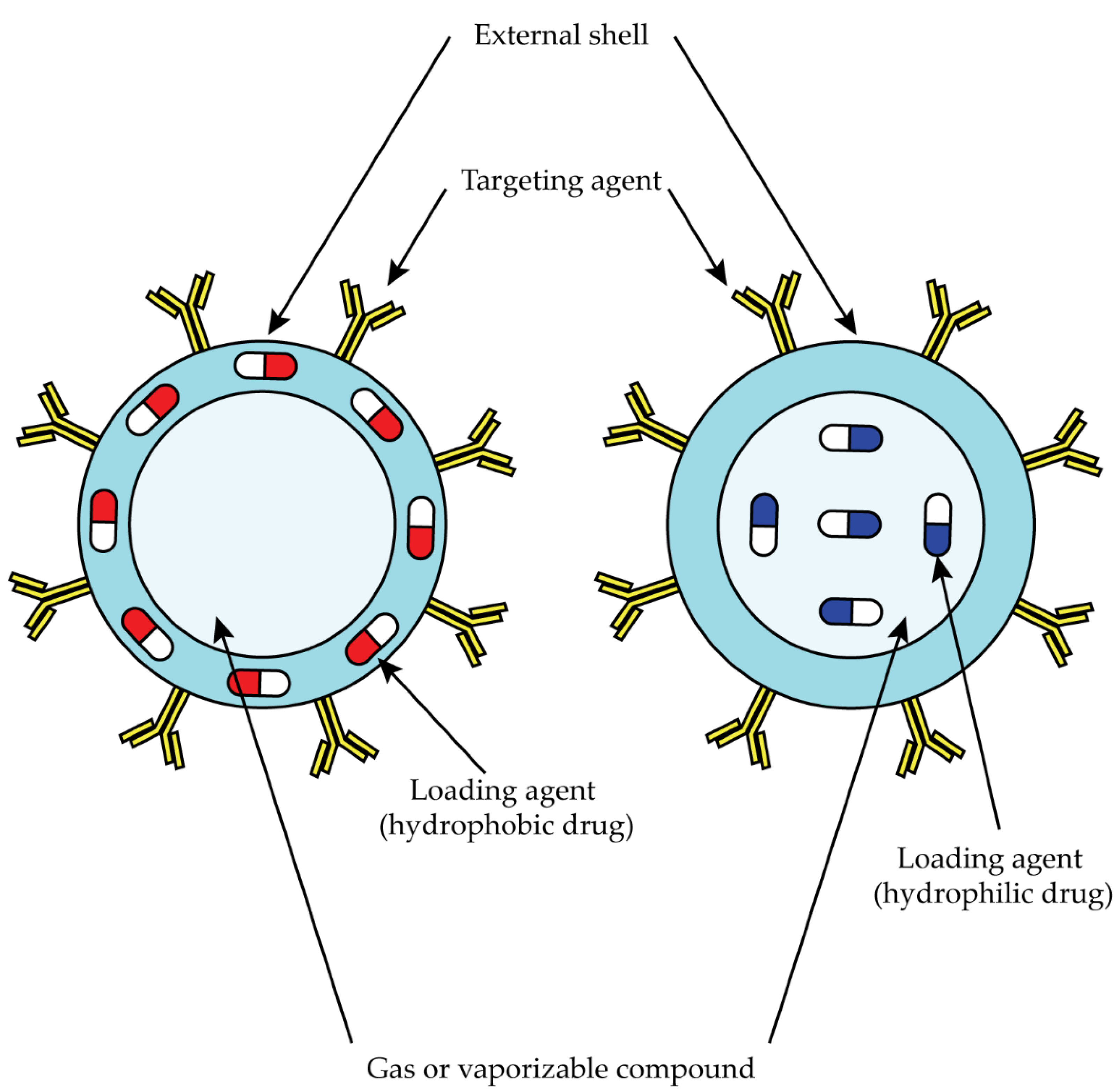
3.5. Nanoparticle-Based Target Therapy
3.6. Anti-GPC1 NPs
3.7. Nanobubbles in PDAC
3.8. Chitosan and Chitosan Nanobubbles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.M.; Thomassian, S.; Gong, J.; Hendifar, A.; Osipov, A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers 2021, 13, 5510. [Google Scholar] [CrossRef] [PubMed]
- Montemagno, C.; Cassim, S.; De Leiris, N.; Durivault, J.; Faraggi, M.; Pagès, G. Pancreatic Ductal Adenocarcinoma: The Dawn of the Era of Nuclear Medicine? Int. J. Mol. Sci. 2021, 22, 6413. [Google Scholar] [CrossRef]
- Aslan, M.; Shahbazi, R.; Ulubayram, K.; Ozpolat, B. Targeted Therapies for Pancreatic Cancer and Hurdles Ahead. Anticancer. Res. 2018, 38, 6591–6606. [Google Scholar] [CrossRef]
- Cancer of the Pancreas—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 30 August 2022).
- Ushio, J.; Kanno, A.; Ikeda, E.; Ando, K.; Nagai, H.; Miwata, T.; Kawasaki, Y.; Tada, Y.; Yokoyama, K.; Numao, N.; et al. Pancreatic Ductal Adenocarcinoma: Epidemiology and Risk Factors. Diagnostics 2021, 11, 562. [Google Scholar] [CrossRef]
- Montemagno, C.; Cassim, S.; Pouyssegur, J.; Broisat, A.; Pagès, G. From Malignant Progression to Therapeutic Targeting: Current Insights of Mesothelin in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 4067. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic Cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic Ductal Adenocarcinoma: Treatment Hurdles, Tumor Microenvironment and Immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic Cancer: A Review of Clinical Diagnosis, Epidemiology, Treatment and Outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic Ductal Adenocarcinoma: Biological Hallmarks, Current Status, and Future Perspectives of Combined Modality Treatment Approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ahuja, N.; Makary, M.A.; Cameron, J.L.; Eckhauser, F.E.; Choti, M.A.; Hruban, R.H.; Pawlik, T.M.; Wolfgang, C.L. 2564 Resected Periampullary Adenocarcinomas at a Single Institution: Trends over Three Decades. HPB 2014, 16, 83–90. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Mitra, S.; Tomar, P.C. Hybridoma Technology; Advancements, Clinical Significance, and Future Aspects. J. Genet. Eng. Biotechnol. 2021, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Hester, R.; Mazur, P.K.; McAllister, F. Immunotherapy in Pancreatic Adenocarcinoma: Beyond “Copy/Paste”. Clin. Cancer Res. 2021, 27, 6287–6297. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Rauth, S.; Aithal, A.; Kaur, S.; Ganguly, K.; Orzechowski, C.; Varshney, G.C.; Jain, M.; Batra, S.K. The Current Landscape of Antibody-Based Therapies in Solid Malignancies. Theranostics 2021, 11, 1493–1512. [Google Scholar] [CrossRef] [PubMed]
- Harada, E.; Serada, S.; Fujimoto, M.; Takahashi, Y.; Takahashi, T.; Hara, H.; Nakatsuka, R.; Sugase, T.; Nishigaki, T.; Saito, Y.; et al. Glypican-1 Targeted Antibody-Based Therapy Induces Preclinical Antitumor Activity against Esophageal Squamous Cell Carcinoma. Oncotarget 2017, 8, 24741–24752. [Google Scholar] [CrossRef]
- Tsujii, S.; Serada, S.; Fujimoto, M.; Uemura, S.; Namikawa, T.; Nomura, T.; Murakami, I.; Hanazaki, K.; Naka, T. Glypican-1 Is a Novel Target for Stroma and Tumor Cell Dual-Targeting Antibody-Drug Conjugates in Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 2495–2505. [Google Scholar] [CrossRef]
- Diaz, K.E.; Lucas, A.L. Familial Pancreatic Ductal Adenocarcinoma. Am. J. Pathol. 2019, 189, 36–43. [Google Scholar] [CrossRef]
- Jenne, D.E.; Reomann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers Syndrome Is Caused by Mutations in a Novel Serine Threoninekinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- DuFort, C.C.; DelGiorno, K.E.; Hingorani, S.R. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2016, 150, 1545–1557.e2. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Van Buren, G.; Fisher, W.E. Pancreatic Cancer: Advances in Treatment. World J. Gastroenterol. 2014, 20, 9354–9360. [Google Scholar] [CrossRef] [PubMed]
- Esposito, I.; Konukiewitz, B.; Schlitter, A.M.; Klöppel, G. Pathology of Pancreatic Ductal Adenocarcinoma: Facts, Challenges and Future Developments. World J. Gastroenterol. 2014, 20, 13833–13841. [Google Scholar] [CrossRef]
- Wang, L.; Xie, D.; Wei, D. Pancreatic Acinar-to-Ductal Metaplasia and Pancreatic Cancer. In Pancreatic Cancer: Methods and Protocols; Su, G.H., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 299–308. ISBN 978-1-4939-8879-2. [Google Scholar]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A Renewed Model of Pancreatic Cancer Evolution Based on Genomic Rearrangement Patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.M.; Mills, K.; Mendonça, S.C.; Abel, G.A.; Basu, B.; Carroll, N.; Ballard, S.; Lancaster, J.; Hamilton, W.; Rubin, G.P.; et al. Symptoms and Patient Factors Associated with Diagnostic Intervals for Pancreatic Cancer (SYMPTOM Pancreatic Study): A Prospective Cohort Study. Lancet Gastroenterol. Hepatol. 2016, 1, 298–306. [Google Scholar] [CrossRef]
- Aslanian, H.R.; Lee, J.H.; Canto, M.I. AGA Clinical Practice Update on Pancreas Cancer Screening in High-Risk Individuals: Expert Review. Gastroenterology 2020, 159, 358–362. [Google Scholar] [CrossRef]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA -Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The Challenge of Drug Resistance in Pancreatic Ductal Adenocarcinoma: A Current Overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef]
- Ansari, D.; Ohlsson, H.; Althini, C.; Bauden, M.; Zhou, Q.; Hu, D.; Andersson, R. The Hippo Signaling Pathway in Pancreatic Cancer. Anticancer Res. 2019, 39, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Braun, L.M.; Lagies, S.; Guenzle, J.; Fichtner-Feigl, S.; Wittel, U.A.; Kammerer, B. Metabolic Adaptation during Nab-Paclitaxel Resistance in Pancreatic Cancer Cell Lines. Cells 2020, 9, 1251. [Google Scholar] [CrossRef] [PubMed]
- Leroux, C.; Konstantinidou, G. Targeted Therapies for Pancreatic Cancer: Overview of Current Treatments and New Opportunities for Personalized Oncology. Cancers 2021, 13, 799. [Google Scholar] [CrossRef]
- Kleeff, J.; Ishiwata, T.; Kumbasar, A.; Friess, H.; Büchler, M.W.; Lander, A.D.; Korc, M. The Cell-Surface Heparan Sulfate Proteoglycan Glypican-1 Regulates Growth Factor Action in Pancreatic Carcinoma Cells and Is Overexpressed in Human Pancreatic Cancer. J. Clin. Investig. 1998, 102, 1662–1673. [Google Scholar] [CrossRef]
- Lu, H.; Niu, F.; Liu, F.; Gao, J.; Sun, Y.; Zhao, X. Elevated Glypican-1 Expression Is Associated with an Unfavorable Prognosis in Pancreatic Ductal Adenocarcinoma. Cancer Med. 2017, 6, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-Y.; Dong, Y.-P.; Sun, X.; Sui, X.; Zhu, H.; Zhao, Y.-Q.; Zhang, Y.-Y.; Mason, C.; Zhu, Q.; Han, S.-X. High Levels of Serum Glypican-1 Indicate Poor Prognosis in Pancreatic Ductal Adenocarcinoma. Cancer Med. 2018, 7, 5525–5533. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 Identifies Cancer Exosomes and Detects Early Pancreatic Cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Shlissel, M.; Berman, B.; Shaoul, E.; Admon, A.; Vlodavsky, I.; Carey, D.J.; Asundi, V.K.; Reich-Slotky, R.; Ron, D. Identification of Glypican as a Dual Modulator of the Biological Activity of Fibroblast Growth Factors. J. Biol. Chem. 1997, 272, 12415–12421. [Google Scholar] [CrossRef] [Green Version]
- Häcker, U.; Nybakken, K.; Perrimon, N. Heparan Sulphate Proteoglycans: The Sweet Side of Development. Nat. Rev. Mol. Cell Biol. 2005, 6, 530–541. [Google Scholar] [CrossRef]
- Traister, A.; Shi, W.; Filmus, J. Mammalian Notum Induces the Release of Glypicans and Other GPI-Anchored Proteins from the Cell Surface. Biochem. J. 2008, 410, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Selleck, S.B. Glypicans: Proteoglycans with a Surprise. J. Clin. Investig. 2001, 108, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Okolicsanyi, R.K.; van Wijnen, A.J.; Cool, S.M.; Stein, G.S.; Griffiths, L.R.; Haupt, L.M. Heparan Sulfate Proteoglycans and Human Breast Cancer Epithelial Cell Tumorigenicity. J. Cell. Biochem. 2014, 115, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef]
- De Cat, B.; David, G. Developmental Roles of the Glypicans. Semin. Cell Dev. Biol. 2001, 12, 117–125. [Google Scholar] [CrossRef]
- Filmus, J. Glypicans in Growth Control and Cancer. Glycobiology 2001, 11, 19R–23R. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Naruo, H.; Yoshitomi, Y.; Munesue, S.; Kiyono, S.; Kusano, Y.; Hashimoto, K.; Yokoi, T.; Nakanishi, H.; Shimizu, S.; et al. Matrix Metalloproteinase-9 Associated with Heparan Sulphate Chains of GPI-Anchored Cell Surface Proteoglycans Mediates Motility of Murine Colon Adenocarcinoma Cells. J. Biochem. 2008, 143, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fu, H.; Hewitt, S.M.; Dimitrov, D.S.; Ho, M. Therapeutically Targeting Glypican-2 via Single-Domain Antibody-Based Chimeric Antigen Receptors and Immunotoxins in Neuroblastoma. Proc. Natl. Acad. Sci. USA 2017, 114, E6623–E6631. [Google Scholar] [CrossRef]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T Cells Redirected to Glypican-3 for the Treatment of Hepatocellular Carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Luo, H.; Fan, M.; Wu, X.; Shi, B.; Di, S.; Liu, Y.; Pan, Z.; Jiang, H.; Li, Z. Development of GPC3-Specific Chimeric Antigen Receptor-Engineered Natural Killer Cells for the Treatment of Hepatocellular Carcinoma. Mol. Ther. 2018, 26, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Gesta, S.; Blüher, M.; Yamamoto, Y.; Norris, A.W.; Berndt, J.; Kralisch, S.; Boucher, J.; Lewis, C.; Kahn, C.R. Evidence for a Role of Developmental Genes in the Origin of Obesity and Body Fat Distribution. Proc. Natl. Acad. Sci. USA 2006, 103, 6676–6681. [Google Scholar] [CrossRef] [PubMed]
- Leelalertlauw, C.; Korwutthikulrangsri, M.; Mahachoklertwattana, P.; Chanprasertyothin, S.; Khlairit, P.; Pongratanakul, S.; Poomthavorn, P. Serum Glypican 4 Level in Obese Children and Its Relation to Degree of Obesity. Clin. Endocrinol. 2017, 87, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Tokunaga, K.; Doi, K.; Fujita, T.; Suzuki, H.; Katoh, T.; Watanabe, T.; Nishida, N.; Mabuchi, A.; Takahashi, A.; et al. Common Variation in GPC5 Is Associated with Acquired Nephrotic Syndrome. Nat. Genet. 2011, 43, 459–463. [Google Scholar] [CrossRef]
- Veugelers, M.; De Cat, B.; Ceulemans, H.; Bruystens, A.M.; Coomans, C.; Dürr, J.; Vermeesch, J.; Marynen, P.; David, G. Glypican-6, a New Member of the Glypican Family of Cell Surface Heparan Sulfate Proteoglycans. J. Biol. Chem. 1999, 274, 26968–26977. [Google Scholar] [CrossRef]
- Zhou, F.; Shang, W.; Yu, X.; Tian, J. Glypican-3: A Promising Biomarker for Hepatocellular Carcinoma Diagnosis and Treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef]
- Truong, Q.; Justiniano, I.O.; Nocon, A.L.; Soon, J.T.; Wissmueller, S.; Campbell, D.H.; Walsh, B.J. Glypican-1 as a Biomarker for Prostate Cancer: Isolation and Characterization. J Cancer 2016, 7, 1002–1009. [Google Scholar] [CrossRef]
- Su, G.; Meyer, K.; Nandini, C.D.; Qiao, D.; Salamat, S.; Friedl, A. Glypican-1 Is Frequently Overexpressed in Human Gliomas and Enhances FGF-2 Signaling in Glioma Cells. Am. J. Pathol. 2006, 168, 2014–2026. [Google Scholar] [CrossRef]
- Huang, G.; Ge, G.; Izzi, V.; Greenspan, D.S. A3 Chains of Type V Collagen Regulate Breast Tumour Growth via Glypican-1. Nat. Commun. 2017, 8, 14351. [Google Scholar] [CrossRef]
- Capurro, M.; Wanless, I.R.; Sherman, M.; Deboer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A Novel Serum and Histochemical Marker for Hepatocellular Carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef]
- Baumhoer, D.; Tornillo, L.; Stadlmann, S.; Roncalli, M.; Diamantis, E.K.; Terracciano, L.M. Glypican 3 Expression in Human Nonneoplastic, Preneoplastic, and Neoplastic Tissues: A Tissue Microarray Analysis of 4,387 Tissue Samples. Am. J. Clin. Pathol. 2008, 129, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Nakatsura, T.; Kageshita, T.; Ito, S.; Wakamatsu, K.; Monji, M.; Ikuta, Y.; Senju, S.; Ono, T.; Nishimura, Y. Identification of Glypican-3 as a Novel Tumor Marker for Melanoma. Clin. Cancer Res. 2004, 10, 6612–6621. [Google Scholar] [CrossRef] [PubMed]
- Stadlmann, S.; Gueth, U.; Baumhoer, D.; Moch, H.; Terracciano, L.; Singer, G. Glypican-3 Expression in Primary and Recurrent Ovarian Carcinomas. Int. J. Gynecol. Pathol. 2007, 26, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Saikali, Z.; Sinnett, D. Expression of Glypican 3 (GPC3) in Embryonal Tumors. Int. J. Cancer 2000, 89, 418–422. [Google Scholar] [CrossRef]
- Shibui, Y.; Miyoshi, K.; Kohashi, K.; Kinoshita, Y.; Kuda, M.; Yamamoto, H.; Taguchi, T.; Oda, Y. Glypican-3 Expression in Malignant Small Round Cell Tumors. Oncol. Lett. 2019, 17, 3523–3528. [Google Scholar] [CrossRef]
- Kolluri, A.; Ho, M. The Role of Glypican-3 in Regulating Wnt, YAP, and Hedgehog in Liver Cancer. Front. Oncol. 2019, 9, 708. [Google Scholar] [CrossRef]
- Yoo, H.J.; Hwang, S.Y.; Cho, G.J.; Hong, H.C.; Choi, H.Y.; Hwang, T.G.; Kim, S.M.; Blüher, M.; Youn, B.-S.; Baik, S.H.; et al. Association of Glypican-4 with Body Fat Distribution, Insulin Resistance, and Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2013, 98, 2897–2901. [Google Scholar] [CrossRef]
- Williamson, D.; Selfe, J.; Gordon, T.; Lu, Y.-J.; Pritchard-Jones, K.; Murai, K.; Jones, P.; Workman, P.; Shipley, J. Role for Amplification and Expression of Glypican-5 in Rhabdomyosarcoma. Cancer Res. 2007, 67, 57–65. [Google Scholar] [CrossRef]
- Li, F.; Shi, W.; Capurro, M.; Filmus, J. Glypican-5 Stimulates Rhabdomyosarcoma Cell Proliferation by Activating Hedgehog Signaling. J. Cell Biol. 2011, 192, 691–704. [Google Scholar] [CrossRef]
- Guo, L.; Wang, J.; Zhang, T.; Yang, Y. Glypican-5 Is a Tumor Suppressor in Non-Small Cell Lung Cancer Cells. Biochem. Biophys. Rep. 2016, 6, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Dinccelik-Aslan, M.; Gumus-Akay, G.; Elhan, A.H.; Unal, E.; Tukun, A. Diagnostic and Prognostic Significance of Glypican 5 and Glypican 6 Gene Expression Levels in Gastric Adenocarcinoma. Mol. Clin. Oncol. 2015, 3, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Litwack, E.D.; Ivins, J.K.; Kumbasar, A.; Paine-Saunders, S.; Stipp, C.S.; Lander, A.D. Expression of the Heparan Sulfate Proteoglycan Glypican-1 in the Developing Rodent. Dev. Dyn. 1998, 211, 72–87. [Google Scholar] [CrossRef]
- Awad, W.; Logan, D.; Mani, K. GPC1 (Glypican 1). Atlas Genet. Cytogenet. Oncol. Haematol. 2014, 18, 461–464. [Google Scholar] [CrossRef]
- Awad, W.; Adamczyk, B.; Örnros, J.; Karlsson, N.G.; Mani, K.; Logan, D.T. Structural Aspects of N-Glycosylations and the C-Terminal Region in Human Glypican-1. J. Biol. Chem. 2015, 290, 22991–23008. [Google Scholar] [CrossRef] [PubMed]
- Svensson, G.; Awad, W.; Håkansson, M.; Mani, K.; Logan, D.T. Crystal Structure of N-Glycosylated Human Glypican-1 Core Protein: Structure of Two Loops Evolutionarily Conserved in Vertebrate Glypican-1. J. Biol. Chem. 2012, 287, 14040–14051. [Google Scholar] [CrossRef]
- Lund, M.E.; Campbell, D.H.; Walsh, B.J. The Role of Glypican-1 in the Tumour Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 163–176. [Google Scholar] [CrossRef]
- Jen, Y.-H.L.; Musacchio, M.; Lander, A.D. Glypican-1 Controls Brain Size through Regulation of Fibroblast Growth Factor Signaling in Early Neurogenesis. Neural Dev. 2009, 4, 33. [Google Scholar] [CrossRef]
- Hara, H.; Takahashi, T.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Nakatsuka, R.; Harada, E.; Nishigaki, T.; Takahashi, Y.; Nojima, S.; et al. Overexpression of Glypican-1 Implicates Poor Prognosis and Their Chemoresistance in Oesophageal Squamous Cell Carcinoma. Br. J. Cancer 2016, 115, 66–75. [Google Scholar] [CrossRef]
- Wilson, N.H.; Stoeckli, E.T. Sonic Hedgehog Regulates Its Own Receptor on Postcrossing Commissural Axons in a Glypican1-Dependent Manner. Neuron 2013, 79, 478–491. [Google Scholar] [CrossRef]
- Watanabe, N.; Araki, W.; Chui, D.-H.; Makifuchi, T.; Ihara, Y.; Tabira, T. Glypican-1 as an Abeta Binding HSPG in the Human Brain: Its Localization in DIG Domains and Possible Roles in the Pathogenesis of Alzheimer’s Disease. FASEB J. 2004, 18, 1013–1015. [Google Scholar] [CrossRef]
- Löfgren, K.; Cheng, F.; Fransson, L.-A.; Bedecs, K.; Mani, K. Involvement of Glypican-1 Autoprocessing in Scrapie Infection. Eur. J. Neurosci. 2008, 28, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Mani, K.; Cheng, F.; Fransson, L.-A. Defective Nitric Oxide-Dependent, Deaminative Cleavage of Glypican-1 Heparan Sulfate in Niemann-Pick C1 Fibroblasts. Glycobiology 2006, 16, 711–718. [Google Scholar] [CrossRef]
- Frampton, A.E.; Prado, M.M.; López-Jiménez, E.; Fajardo-Puerta, A.B.; Jawad, Z.A.R.; Lawton, P.; Giovannetti, E.; Habib, N.A.; Castellano, L.; Stebbing, J.; et al. Glypican-1 Is Enriched in Circulating-Exosomes in Pancreatic Cancer and Correlates with Tumor Burden. Oncotarget 2018, 9, 19006–19013. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, T.; Whipple, C.A.; Lopez, M.E.; Gunn, J.; Young, A.; Lander, A.D.; Korc, M. Glypican-1 Modulates the Angiogenic and Metastatic Potential of Human and Mouse Cancer Cells. J. Clin. Investig. 2008, 118, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Ishikawa, S.; Ushiku, T.; Morikawa, T.; Isagawa, T.; Yamagishi, M.; Yamamoto, H.; Katoh, H.; Takeshita, K.; Arita, J.; et al. EVI1 Modulates Oncogenic Role of GPC1 in Pancreatic Carcinogenesis. Oncotarget 2017, 8, 99552–99566. [Google Scholar] [CrossRef]
- Li, J.; Kleeff, J.; Kayed, H.; Felix, K.; Penzel, R.; Büchler, M.W.; Korc, M.; Friess, H. Glypican-1 Antisense Transfection Modulates TGF-Beta-Dependent Signaling in Colo-357 Pancreatic Cancer Cells. Biochem. Biophys. Res. Commun. 2004, 320, 1148–1155. [Google Scholar] [CrossRef]
- Kayed, H.; Kleeff, J.; Keleg, S.; Jiang, X.; Penzel, R.; Giese, T.; Zentgraf, H.; Büchler, M.W.; Korc, M.; Friess, H. Correlation of Glypican-1 Expression with TGF-Beta, BMP, and Activin Receptors in Pancreatic Ductal Adenocarcinoma. Int. J. Oncol. 2006, 29, 1139–1148. [Google Scholar]
- Kansara, R.R.; Speziali, C. Immunotherapy in Hematologic Malignancies. Curr. Oncol. 2020, 27, S124–S131. [Google Scholar] [CrossRef]
- Sabanathan, D.; Campbell, D.H.; Velonas, V.M.; Wissmueller, S.; Mazure, H.; Trifunovic, M.; Poursoltan, P.; Ho Shon, K.; Mackay, T.R.; Lund, M.E.; et al. Safety and Tolerability of Miltuximab®—A First in Human Study in Patients with Advanced Solid Cancers. Asia Ocean. J. Nucl. Med. Biol. 2021, 9, 86–100. [Google Scholar] [CrossRef]
- Ghosh, S.; Huda, P.; Fletcher, N.; Campbell, D.; Thurecht, K.J.; Walsh, B. Clinical Development of an Anti-GPC-1 Antibody for the Treatment of Cancer. Expert Opin. Biol. Ther. 2022, 22, 603–613. [Google Scholar] [CrossRef]
- Busato, D.; Mossenta, M.; Baboci, L.; Di Cintio, F.; Toffoli, G.; Dal Bo, M. Novel Immunotherapeutic Approaches for Hepatocellular Carcinoma Treatment. Expert Rev. Clin. Pharmacol. 2019, 12, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T Design: Elements and Their Synergistic Function. eBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Zhou, J.; Chen, R.; Jin, Y.; Ni, T.; Qian, L.; Xiao, C. Cluster of Differentiation 19 Chimeric Antigen Receptor T-Cell Therapy in Pediatric Acute Lymphoblastic Leukemia. Oncol. Lett. 2020, 20, 36. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Kato, D.; Yaguchi, T.; Iwata, T.; Katoh, Y.; Morii, K.; Tsubota, K.; Takise, Y.; Tamiya, M.; Kamada, H.; Akiba, H.; et al. GPC1 Specific CAR-T Cells Eradicate Established Solid Tumor without Adverse Effects and Synergize with Anti-PD-1 Ab. eLife 2020, 9, e49392. [Google Scholar] [CrossRef]
- Munekage, E.; Serada, S.; Tsujii, S.; Yokota, K.; Kiuchi, K.; Tominaga, K.; Fujimoto, M.; Kanda, M.; Uemura, S.; Namikawa, T.; et al. A Glypican-1-Targeted Antibody-Drug Conjugate Exhibits Potent Tumor Growth Inhibition in Glypican-1-Positive Pancreatic Cancer and Esophageal Squamous Cell Carcinoma. Neoplasia 2021, 23, 939–950. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Ford, C.H.; Newman, C.E.; Johnson, J.R.; Woodhouse, C.S.; Reeder, T.A.; Rowland, G.F.; Simmonds, R.G. Localisation and Toxicity Study of a Vindesine-Anti-CEA Conjugate in Patients with Advanced Cancer. Br. J. Cancer 1983, 47, 35–42. [Google Scholar] [CrossRef]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab Vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients With Relapsed or Refractory Hodgkin’s Lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef]
- Burris, H.A.; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II Study of the Antibody Drug Conjugate Trastuzumab-DM1 for the Treatment of Human Epidermal Growth Factor Receptor 2 (HER2)-Positive Breast Cancer after Prior HER2-Directed Therapy. J. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab Emtansine: A Unique Antibody-Drug Conjugate in Development for Human Epidermal Growth Factor Receptor 2-Positive Cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef]
- Krop, I.E.; LoRusso, P.; Miller, K.D.; Modi, S.; Yardley, D.; Rodriguez, G.; Guardino, E.; Lu, M.; Zheng, M.; Girish, S.; et al. A Phase II Study of Trastuzumab Emtansine in Patients with Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Who Were Previously Treated with Trastuzumab, Lapatinib, an Anthracycline, a Taxane, and Capecitabine. J. Clin. Oncol. 2012, 30, 3234–3241. [Google Scholar] [CrossRef]
- Goy, A.; Forero, A.; Wagner-Johnston, N.; Christopher Ehmann, W.; Tsai, M.; Hatake, K.; Ananthakrishnan, R.; Volkert, A.; Vandendries, E.; Ogura, M. A Phase 2 Study of Inotuzumab Ozogamicin in Patients with Indolent B-Cell Non-Hodgkin Lymphoma Refractory to Rituximab Alone, Rituximab and Chemotherapy, or Radioimmunotherapy. Br. J. Haematol. 2016, 174, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Tobinai, K.; Hatake, K.; Davies, A.; Crump, M.; Ananthakrishnan, R.; Ishibashi, T.; Paccagnella, M.L.; Boni, J.; Vandendries, E.; et al. Phase I Study of Inotuzumab Ozogamicin Combined with R-CVP for Relapsed/Refractory CD22+ B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2016, 22, 4807–4816. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, T.; Takahashi, T.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Hara, H.; Sugase, T.; Otsuru, T.; Saito, Y.; Tsujii, S.; et al. Anti-Glypican-1 Antibody–Drug Conjugate Is a Potential Therapy against Pancreatic Cancer. Br. J. Cancer 2020, 122, 1333–1341. [Google Scholar] [CrossRef]
- Tran, S.; DeGiovanni, P.-J.; Piel, B.; Rai, P. Cancer Nanomedicine: A Review of Recent Success in Drug Delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle Therapeutics: An Emerging Treatment Modality for Cancer. Nat. Rev. Drug. Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 12320–12364. [Google Scholar] [CrossRef]
- Bhatia, S. Nanoparticles Types, Classification, Characterization, Fabrication Methods and Drug Delivery Applications. In Natural Polymer Drug Delivery Systems: Nanoparticles, Plants, and Algae; Bhatia, S., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 33–93. ISBN 978-3-319-41129-3. [Google Scholar]
- Yadav, K.S.; Mishra, D.K.; Deshpande, A.; Pethe, A.M. Chapter 7—Levels of Drug Targeting. In Basic Fundamentals of Drug Delivery; Tekade, R.K., Ed.; Advances in Pharmaceutical Product Development and Research; Academic Press: Cambridge, MA, USA, 2019; pp. 269–305. ISBN 978-0-12-817909-3. [Google Scholar]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Thomas, A.G.; Awasthi, N. Targeted Therapy for Pancreatic Cancer: Lessons Learned and Future Opportunities. Dig. Med. Res. 2021, 4, 32. [Google Scholar] [CrossRef]
- Markowski, A.; Migdał, P.; Zygmunt, A.; Zaremba-Czogalla, M.; Gubernator, J. Evaluation of the In Vitro Cytotoxic Activity of Ursolic Acid PLGA Nanoparticles against Pancreatic Ductal Adenocarcinoma Cell Lines. Materials 2021, 14, 4917. [Google Scholar] [CrossRef]
- Ray, P.; Dutta, D.; Haque, I.; Nair, G.; Mohammed, J.; Parmer, M.; Kale, N.; Orr, M.; Jain, P.; Banerjee, S.; et al. PH-Sensitive Nanodrug Carriers for Codelivery of ERK Inhibitor and Gemcitabine Enhance the Inhibition of Tumor Growth in Pancreatic Cancer. Mol. Pharm. 2021, 18, 87–100. [Google Scholar] [CrossRef]
- Zhao, X.; Li, F.; Li, Y.; Wang, H.; Ren, H.; Chen, J.; Nie, G.; Hao, J. Co-Delivery of HIF1α SiRNA and Gemcitabine via Biocompatible Lipid-Polymer Hybrid Nanoparticles for Effective Treatment of Pancreatic Cancer. Biomaterials 2015, 46, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Li, J.; Wang, Y.; Qian, C.; Chen, Y.; Zhang, Q.; Wu, W.; Lin, Z.; Liang, J.; Shuai, X.; et al. Combination of SiRNA-Directed Kras Oncogene Silencing and Arsenic-Induced Apoptosis Using a Nanomedicine Strategy for the Effective Treatment of Pancreatic Cancer. Nanomedicine 2014, 10, 463–472. [Google Scholar] [CrossRef]
- Jung, J.Y.; Ryu, H.J.; Lee, S.-H.; Kim, D.-Y.; Kim, M.J.; Lee, E.J.; Ryu, Y.-M.; Kim, S.-Y.; Kim, K.-P.; Choi, E.Y.; et al. SiRNA Nanoparticle Targeting PD-L1 Activates Tumor Immunity and Abrogates Pancreatic Cancer Growth in Humanized Preclinical Model. Cells 2021, 10, 2734. [Google Scholar] [CrossRef]
- Confeld, M.I.; Mamnoon, B.; Feng, L.; Jensen-Smith, H.; Ray, P.; Froberg, J.; Kim, J.; Hollingsworth, M.A.; Quadir, M.; Choi, Y.; et al. Targeting the Tumor Core: Hypoxia-Responsive Nanoparticles for the Delivery of Chemotherapy to Pancreatic Tumors. Mol. Pharm. 2020, 17, 2849–2863. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Wang, L.-Y.; Li, S.-J.; Tong, T.; Wang, L.; Huang, C.-S.; Xu, Q.-C.; Huang, X.-T.; Li, J.-H.; Wu, J.; et al. Histone Methyltransferase G9a Inhibitor-Loaded Redox-Responsive Nanoparticles for Pancreatic Ductal Adenocarcinoma Therapy. Nanoscale 2020, 12, 15767–15774. [Google Scholar] [CrossRef]
- Qiu, W.; Chen, R.; Chen, X.; Zhang, H.; Song, L.; Cui, W.; Zhang, J.; Ye, D.; Zhang, Y.; Wang, Z. Oridonin-Loaded and GPC1-Targeted Gold Nanoparticles for Multimodal Imaging and Therapy in Pancreatic Cancer. Int. J. Nanomed. 2018, 13, 6809–6827. [Google Scholar] [CrossRef]
- Martins Estevão, B.; José Comparetti, E.; Cristina Rissi, N.; Zucolotto, V. Anti-GPC1-Modified Mesoporous Silica Nanoparticles as Nanocarriers for Combination Therapy and Targeting of PANC-1 Cells. Mater. Adv. 2021, 2, 5224–5235. [Google Scholar] [CrossRef]
- Zhao, Y.-Z.; Du, L.-N.; Lu, C.-T.; Jin, Y.-G.; Ge, S.-P. Potential and Problems in Ultrasound-Responsive Drug Delivery Systems. Int. J. Nanomed. 2013, 8, 1621–1633. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Yang, L.; Jin, J.; Yang, F.; Liu, D.; Hu, K.; Wang, Q.; Yue, Y.; Gu, N. Micro/Nano-Bubble-Assisted Ultrasound to Enhance the EPR Effect and Potential Theranostic Applications. Theranostics 2020, 10, 462–483. [Google Scholar] [CrossRef] [PubMed]
- Argenziano, M.; Arpicco, S.; Brusa, P.; Cavalli, R.; Chirio, D.; Dosio, F.; Gallarate, M.; Peira, E.; Stella, B.; Ugazio, E. Developing Actively Targeted Nanoparticles to Fight Cancer: Focus on Italian Research. Pharmaceutics 2021, 13, 1538. [Google Scholar] [CrossRef] [PubMed]
- Marano, F.; Frairia, R.; Rinella, L.; Argenziano, M.; Bussolati, B.; Grange, C.; Mastrocola, R.; Castellano, I.; Berta, L.; Cavalli, R.; et al. Combining Doxorubicin-Nanobubbles and Shockwaves for Anaplastic Thyroid Cancer Treatment: Preclinical Study in a Xenograft Mouse Model. Endocr. Relat. Cancer 2017, 24, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Marano, F.; Argenziano, M.; Frairia, R.; Adamini, A.; Bosco, O.; Rinella, L.; Fortunati, N.; Cavalli, R.; Catalano, M.G. Doxorubicin-Loaded Nanobubbles Combined with Extracorporeal Shock Waves: Basis for a New Drug Delivery Tool in Anaplastic Thyroid Cancer. Thyroid 2016, 26, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhao, P.; Zhou, Y.; Li, Q.; Cai, W.; Zhao, Z.; Shen, J.; Yao, K.; Duan, Y. Preparation of Multifunctional Nanobubbles and Their Application in Bimodal Imaging and Targeted Combination Therapy of Early Pancreatic Cancer. Sci. Rep. 2021, 11, 6254. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.; McEwan, C.; Nesbitt, H.; Bovornchutichai, P.; Averre, R.; Borden, M.; McHale, A.P.; Callan, J.F.; Stride, E. Reducing Tumour Hypoxia via Oral Administration of Oxygen Nanobubbles. PLoS ONE 2016, 11, e0168088. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, A.; Banerjee, R. Nanobubble Liposome Complexes for Diagnostic Imaging and Ultrasound-Triggered Drug Delivery in Cancers: A Theranostic Approach. ACS Omega 2019, 4, 15567–15580. [Google Scholar] [CrossRef]
- Rizeq, B.R.; Younes, N.N.; Rasool, K.; Nasrallah, G.K. Synthesis, Bioapplications, and Toxicity Evaluation of Chitosan-Based Nanoparticles. Int. J. Mol. Sci. 2019, 20, E5776. [Google Scholar] [CrossRef]
- Wang, W.; Meng, Q.; Li, Q.; Liu, J.; Zhou, M.; Jin, Z.; Zhao, K. Chitosan Derivatives and Their Application in Biomedicine. Int. J. Mol. Sci. 2020, 21, E487. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, E53. [Google Scholar] [CrossRef] [Green Version]
- Aibani, N.; Rai, R.; Patel, P.; Cuddihy, G.; Wasan, E.K. Chitosan Nanoparticles at the Biological Interface: Implications for Drug Delivery. Pharmaceutics 2021, 13, 1686. [Google Scholar] [CrossRef]
- Zhou, X.; Guo, L.; Shi, D.; Duan, S.; Li, J. Biocompatible Chitosan Nanobubbles for Ultrasound-Mediated Targeted Delivery of Doxorubicin. Nanoscale Res. Lett. 2019, 14, 24. [Google Scholar] [CrossRef]
- Cavalli, R.; Bisazza, A.; Trotta, M.; Argenziano, M.; Lembo, D.; Civra, A.; Donalisio, M. New Chitosan Nanobubbles for Ultrasound-Mediated Gene Delivery: Preparation and in Vitro Characterization. Int. J. Nanomed. 2012, 3309. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Argenziano, M.; Marsollier, A.C.; Mariot, V.; Rossi, D.; Selvatici, R.; Dumonceaux, J.; Cavalli, R.; Ferlini, A. Chitosan-Shelled Nanobubbles Irreversibly Encapsulate Morpholino Conjugate Antisense Oligonucleotides and Are Ineffective for Phosphorodiamidate Morpholino-Mediated Gene Silencing of DUX4. Nucleic Acid Ther. 2020, 31, 201–207. [Google Scholar] [CrossRef]
- Argenziano, M.; Bessone, F.; Dianzani, C.; Cucci, M.A.; Grattarola, M.; Pizzimenti, S.; Cavalli, R. Ultrasound-Responsive Nrf2-Targeting SiRNA-Loaded Nanobubbles for Enhancing the Treatment of Melanoma. Pharmaceutics 2022, 14, 341. [Google Scholar] [CrossRef]
- Shen, S.; Li, Y.; Xiao, Y.; Zhao, Z.; Zhang, C.; Wang, J.; Li, H.; Liu, F.; He, N.; Yuan, Y.; et al. Folate-Conjugated Nanobubbles Selectively Target and Kill Cancer Cells via Ultrasound-Triggered Intracellular Explosion. Biomaterials 2018, 181, 293–306. [Google Scholar] [CrossRef]
- Gaitanis, A.; Staal, S. Liposomal Doxorubicin and Nab-Paclitaxel: Nanoparticle Cancer Chemotherapy in Current Clinical Use. Methods Mol. Biol. 2010, 624, 385–392. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Extent | Localized | Advanced | ||
|---|---|---|---|---|
| Major vasculature involvement | Uninvolved or abutted | Uninvolved or abutted | Encased | Distant metastasis, irrespective of the major vascular involvement |
| Clinical stage | Resectable | Borderline resectable | Locally advanced | Metastatic |
| Prevalence of pancreatic cancer among patients newly diagnosed with PDAC, % | 10–15 | 30–35 | 30–35 | 50–55 |
| American Joint Committee on Cancer tumor, node, and metastasis stage | I–II | II–III | II–III | IV |
| Treatment intent | Curative | Curative | Supportive and palliative | Supportive and palliative |
| Treatment | Surgery plus adjuvant systemic therapy | Neoadjuvant systemic therapy; Surgery for resectable patients from favorable response; radiation for unresectable patients without distant metastasis | Neoadjuvant systemic therapy; Surgery for resectable patients from favorable response; radiation for unresectable patients without distant metastasis | Systemic therapy |
| 5-years survival rate, % | 35–45 | 10–15 | 10–15 | <5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busato, D.; Mossenta, M.; Dal Bo, M.; Macor, P.; Toffoli, G. The Proteoglycan Glypican-1 as a Possible Candidate for Innovative Targeted Therapeutic Strategies for Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2022, 23, 10279. https://doi.org/10.3390/ijms231810279
Busato D, Mossenta M, Dal Bo M, Macor P, Toffoli G. The Proteoglycan Glypican-1 as a Possible Candidate for Innovative Targeted Therapeutic Strategies for Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2022; 23(18):10279. https://doi.org/10.3390/ijms231810279
Chicago/Turabian StyleBusato, Davide, Monica Mossenta, Michele Dal Bo, Paolo Macor, and Giuseppe Toffoli. 2022. "The Proteoglycan Glypican-1 as a Possible Candidate for Innovative Targeted Therapeutic Strategies for Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 23, no. 18: 10279. https://doi.org/10.3390/ijms231810279