

Novel Nanotechnology Approaches to Overcome Drug Resistance in the Treatment of Hepatocellular Carcinoma: Glypican 3 as a Useful Target for Innovative Therapies

Abstract
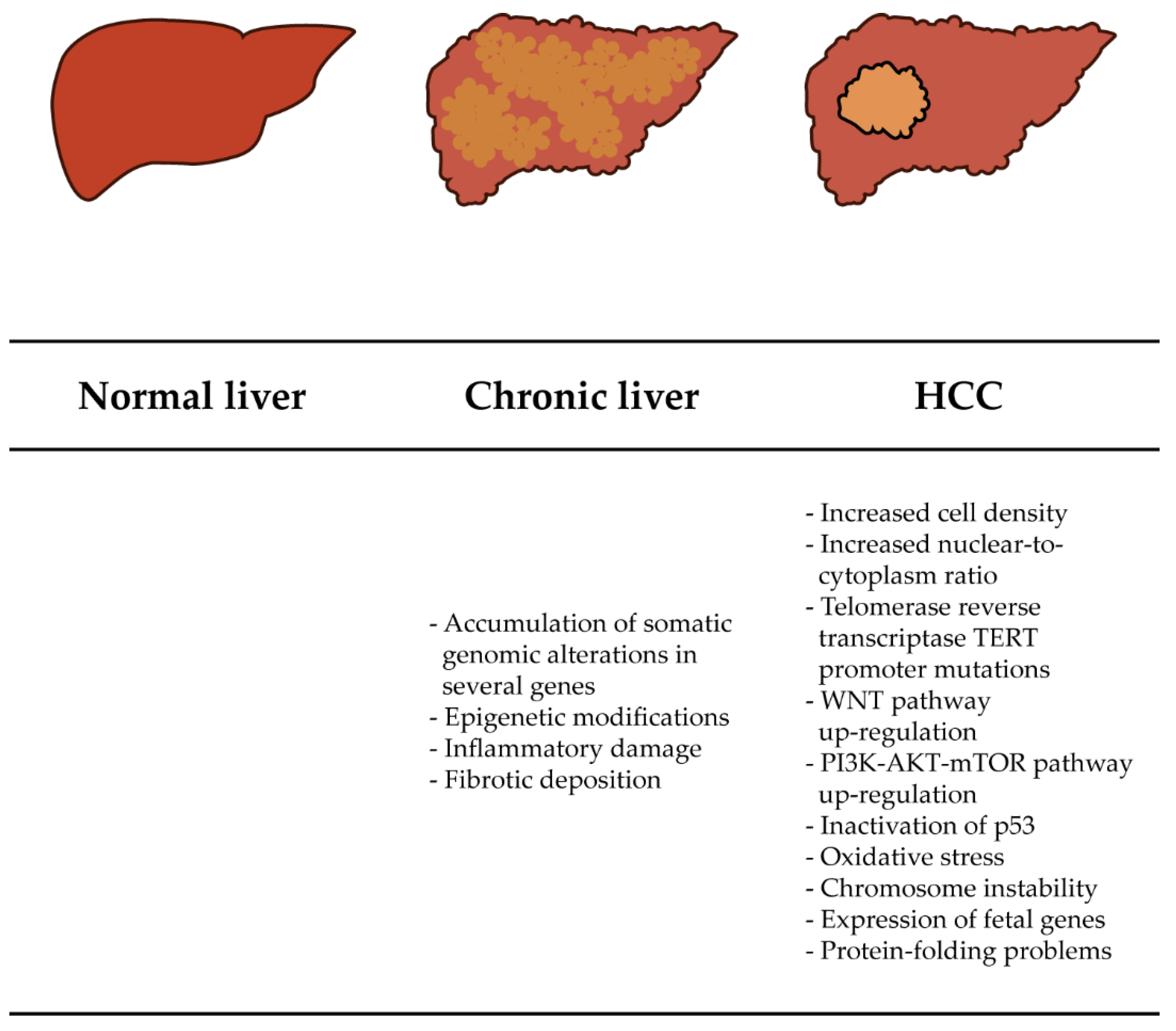
:1. Introduction
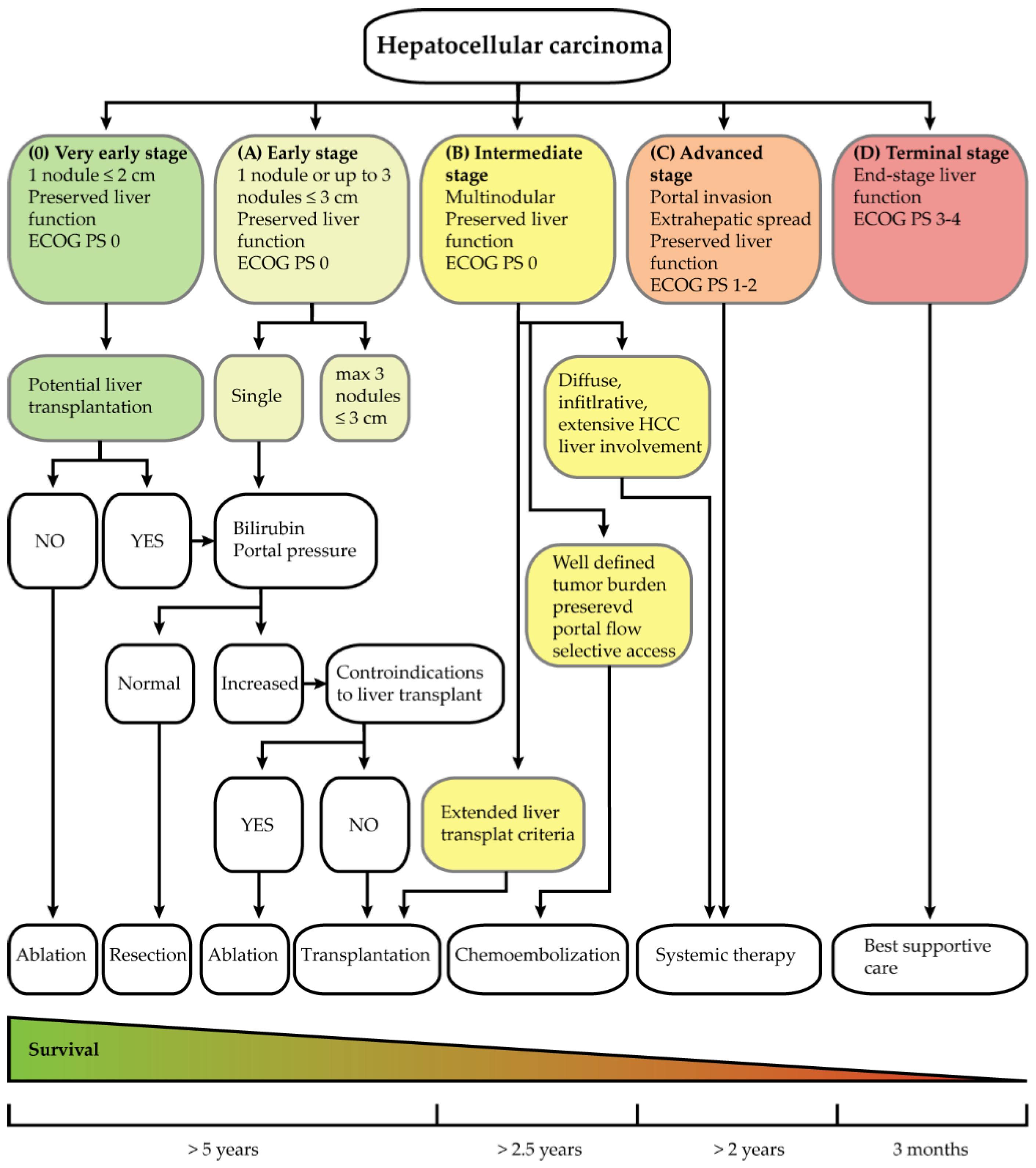
1.1. Diagnosis of HCC
1.2. HCC Treatment
2. Tumor-Associated Antigens Useful for HCC Treatment
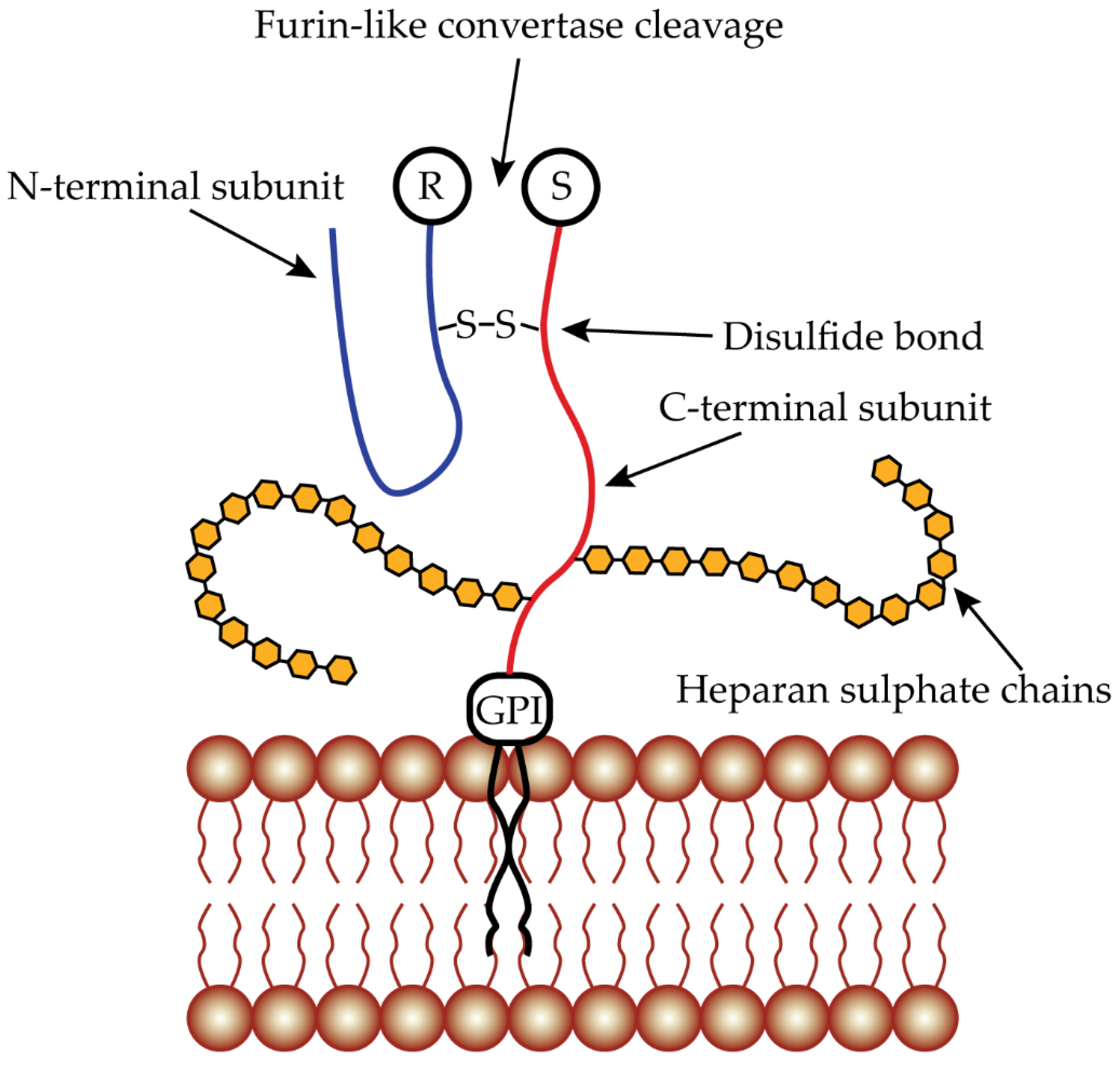
3. Glypican 3 as Targeting Protein
3.1. GPC3 mRNA and Protein Expression
3.2. Antibodies Targeting GPC3
3.2.1. Condrituzumab (GC33)
3.2.2. YP7
3.2.3. G12
3.2.4. Single-Domain Antibodies
3.2.5. Bispecific Antibodies
3.2.6. Anti-GPC3 Immunotoxins
3.2.7. CAR-T Targeting GPC3
3.3. Anti-GPC3 Peptides
3.4. Peptide Vaccines
3.5. Radiopharmaceutical Therapy
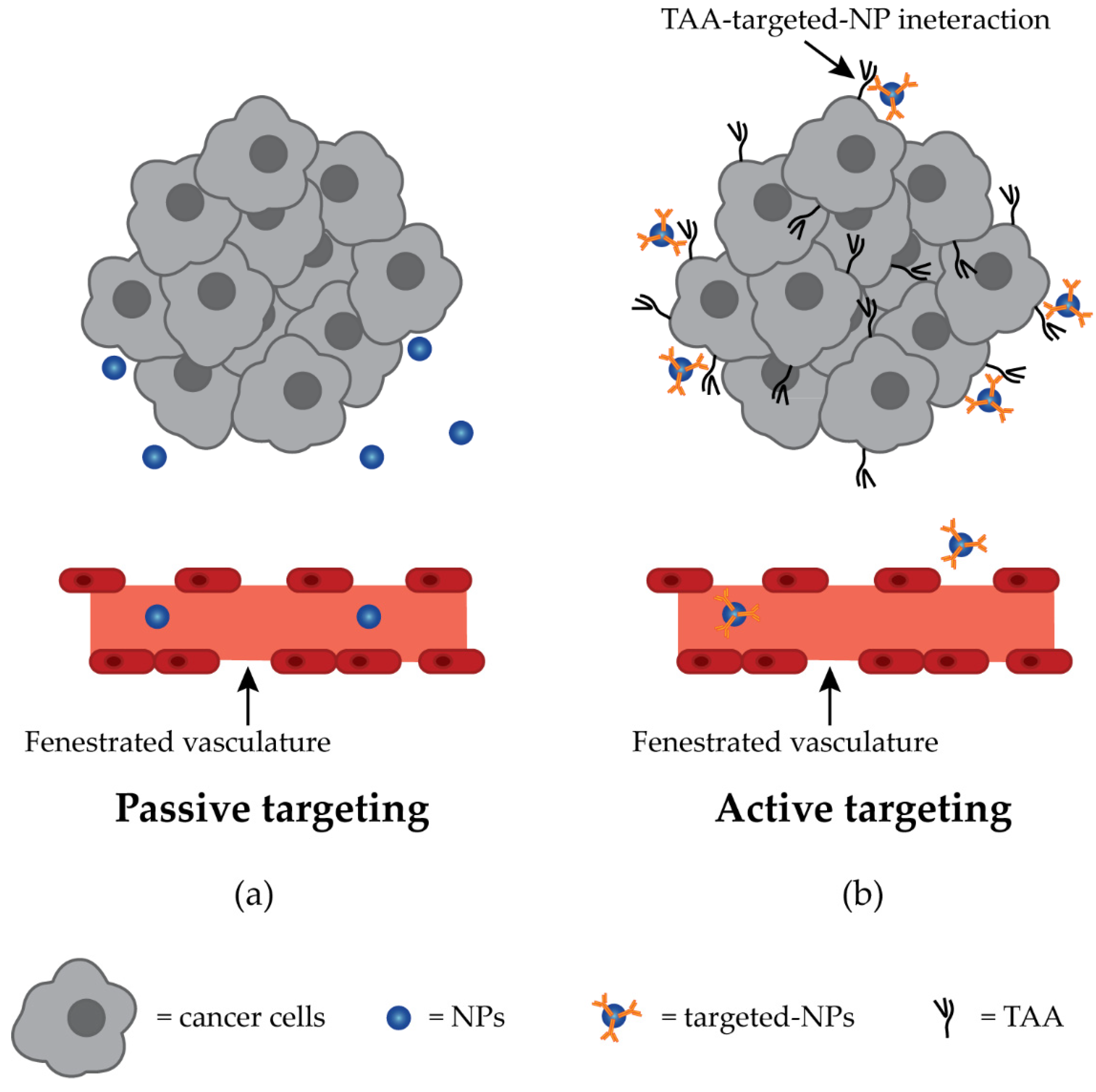
4. Nanoparticles Composed of Organic-Based Nanomaterials
4.1. The Most Common Matrices
4.2. Size and Surface Characteristics
4.3. Drug Encapsulation and Release
4.4. Targeting and Anti-GPC3 NPs Drug Delivery
4.4.1. Polymeric NPs
4.4.2. Liposomes
4.4.3. Imaging
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Petrick, J.L.; London, W.T. Global Epidemiology of Hepatocellular Carcinoma. Clin. Liver Dis. 2015, 19, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Samant, H.; Amiri, H.S.; Zibari, G.B. Addressing the worldwide hepatocellular carcinoma: Epidemiology, prevention and management. J. Gastrointest. Oncol. 2021, 12, S361–S373. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef]
- Huang, W.; Skanderup, A.J.; Lee, C.G. Advances in genomic hepatocellular carcinoma research. GigaScience 2018, 7, giy135. [Google Scholar] [CrossRef]
- Rebouissou, S.; Nault, J.-C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics of hepatocellular carcinoma. Clin. Transl. Med. 2019, 8, 13. [Google Scholar] [CrossRef]
- Renne, S.L.; Sarcognato, S.; Sacchi, D.; Guido, M.; Roncalli, M.; Terracciano, L.; Di Tommaso, L. Hepatocellular carcinoma: A clinical and pathological overview. Pathologica 2021, 113, 203–217. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation Barcelona Clinic Liver Cancer (BCLC) staging system: The 2022 update. J. Hepatol. 2021, 76, 681–693. [Google Scholar] [CrossRef]
- Lune, P.V.; Aal, A.K.A.; Klimkowski, S.; Zarzour, J.G.; Gunn, A.J. Hepatocellular Carcinoma: Diagnosis, Treatment Algorithms, and Imaging Appearance after Transarterial Chemoembolization. J. Clin. Transl. Hepatol. 2017, 6, 175–188. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Gacche, R.N.; Assaraf, Y.G. Redundant angiogenic signaling and tumor drug resistance. Drug Resist. Updates 2018, 36, 47–76. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gašparović, A.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updates 2019, 49, 100670. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Herraez, E.; Lozano, E.; Macias, R.I.R.; Briz, O. Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers 2019, 11, 1677. [Google Scholar] [CrossRef]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updates 2015, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; Pang, B.; Assaraf, Y.G.; Neefjes, J. Old drugs, novel ways out: Drug resistance toward cytotoxic chemotherapeutics. Drug Resist. Updates 2016, 28, 65–81. [Google Scholar] [CrossRef]
- Zhang, J.; Song, Q.; Wu, M.; Zheng, W. The Emerging Roles of Exosomes in the Chemoresistance of Hepatocellular Carcinoma. Curr. Med. Chem. 2020, 28, 93–109. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Leamon, C.P.; Reddy, J.A. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist. Updates 2014, 17, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bar-Zeev, M.; Livney, Y.D.; Assaraf, Y.G. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist. Updates 2017, 31, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updates 2019, 49, 100671. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updates 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Duan, B.; Huang, C.; Bai, J.; Zhang, Y.L.; Wang, X.; Yang, J.; Li, J. Multidrug resistance in hepatocellular carcinoma. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019; ISBN 9780994438188. [Google Scholar]
- Jiang, W.; Xia, J.; Xie, S.; Zou, R.; Pan, S.; Wang, Z.-W.; Assaraf, Y.G.; Zhu, X. Long non-coding RNAs as a determinant of cancer drug resistance: Towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist. Updates 2020, 50, 100683. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.-H.; Chen, Z.-S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updates 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Livney, Y.D.; Assaraf, Y.G. Rationally designed nanovehicles to overcome cancer chemoresistance. Adv. Drug Deliv. Rev. 2013, 65, 1716–1730. [Google Scholar] [CrossRef]
- Narayanan, S.; Cai, C.Y.; Assaraf, Y.G.; Guo, H.Q.; Cui, Q.; Wei, L.; Huang, J.J.; Ashby, C.R., Jr.; Chen, Z.S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updates 2020, 48, 100663. [Google Scholar] [CrossRef]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: An insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biol. Targets Ther. 2019, 13, 33–51. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nanosci. Technol. 2009, 7, 239–250. [Google Scholar] [CrossRef]
- Gavas, S.; Quazi, S.; Karpiński, T.M. Nanoparticles for Cancer Therapy: Current Progress and Challenges. Nanoscale Res. Lett. 2021, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew. Chem. Int. Ed. 2014, 53, 12320–12364. [Google Scholar] [CrossRef]
- Tran, S.; DeGiovanni, P.-J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef]
- Filmus, J.; Capurro, M.; Rast, J. Glypicans. Genome Biol. 2008, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Selleck, S.B. Glypicans: Proteoglycans with a surprise. J. Clin. Investig. 2001, 108, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Baumhoer, D.; Tornillo, L.; Stadlmann, S.; Roncalli, M.; Diamantis, E.K.; Terracciano, L.M. Glypican 3 Expression in Human Nonneoplastic, Preneoplastic, and Neoplastic Tissues. Am. J. Clin. Pathol. 2008, 129, 899–906. [Google Scholar] [CrossRef]
- Yamauchi, N.; Watanabe, A.; Hishinuma, M.; Ohashi, K.-I.; Midorikawa, Y.; Morishita, Y.; Niki, T.; Shibahara, J.; Mori, M.; Makuuchi, M.; et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod. Pathol. 2005, 18, 1591–1598. [Google Scholar] [CrossRef]
- Hsu, H.C.; Cheng, W.; Lai, P.L. Cloning and expression of a developmentally regulated transcript MXR7 in hepatocellular carcinoma: Biological significance and temporospatial distribution. Cancer Res. 1997, 57, 5179–5184. [Google Scholar]
- Zhu, Z.-W.; Friess, H.; Wang, L.; Abou-Shady, M.; Zimmermann, A.; Lander, A.D.; Korc, M.; Kleeff, J.; Büchler, M.W. Enhanced glypican-3 expression differentiates the majority of hepatocellular carcinomas from benign hepatic disorders. Gut 2001, 48, 558–564. [Google Scholar] [CrossRef]
- Nakatsura, T.; Yoshitake, Y.; Senju, S.; Monji, M.; Komori, H.; Motomura, Y.; Hosaka, S.; Beppu, T.; Ishiko, T.; Kamohara, H.; et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem. Biophys. Res. Commun. 2003, 306, 16–25. [Google Scholar] [CrossRef]
- Capurro, M.; Wanless, I.R.; Sherman, M.; Deboer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef]
- Abdul-Al, H.M.; Makhlouf, H.R.; Wang, G.; Goodman, Z.D. Glypican-3 expression in benign liver tissue with active hepatitis C: Implications for the diagnosis of hepatocellular carcinoma. Hum. Pathol. 2008, 39, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-P.; Ariizumi, S.-I.; Nakano, M.; Yamamoto, M. Positive glypican-3 expression in early hepatocellular carcinoma predicts recurrence after hepatectomy. J. Gastroenterol. 2013, 49, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, P.; Zhai, Y.; Qu, C.-F.; Zhang, L.-J.; Tan, Y.-F.; Li, N.; Ding, H.-G. Diagnostic value of glypican-3 in serum and liver for primary hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 4410–4415. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Degos, F.; Dubois, S.; Tessiore, S.; Allegretta, M.; Guttmann, R.D.; Jothy, S.; Belghiti, J.; Bedossa, P.; Paradis, V. Glypican-3 expression in hepatocellular tumors: Diagnostic value for preneoplastic lesions and hepatocellular carcinomas. Hum. Pathol. 2006, 37, 1435–1441. [Google Scholar] [CrossRef]
- Shirakawa, H.; Suzuki, H.; Shimomura, M.; Kojima, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Konishi, M.; Kobayashi, N.; Kinoshita, T.; et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009, 100, 1403–1407. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Zhu, Z.-J.; Teng, D.-H.; Yao, Z.; Gao, W.; Shen, Z.-Y. Glypican-3 expression and its relationship with recurrence of HCC after liver transplantation. World J. Gastroenterol. 2012, 18, 2408–2414. [Google Scholar] [CrossRef]
- Feynman, R. There’s Plenty of Room at the Bottom. Available online: http://www.zyvex.com/nanotech/feynman.html (accessed on 3 May 2019).
- Luby, Š.; Lubyová, M.; Šiffalovič, P.; Jergel, M.; Majková, E. A brief history of nanoscience and foresight in nanotechnology. In Nanomaterials and Nanoarchitectures; Bardosova, M., Wagner, T., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 63–86. ISBN 978-94-017-9920-1. [Google Scholar]
- Jeevanandam, J.; Barhoum, A.; Chan, Y.S.; Dufresne, A.; Danquah, M.K. Review on nanoparticles and nanostructured materials: History, sources, toxicity and regulations. Beilstein J. Nanotechnol. 2018, 9, 1050–1074. [Google Scholar] [CrossRef]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef]
- Hartshorn, C.M.; Bradbury, M.S.; Lanza, G.M.; Nel, A.E.; Rao, J.; Wang, A.; Wiesner, U.B.; Yang, L.; Grodzinski, P. Nanotechnology Strategies to Advance Outcomes in Clinical Cancer Care. ACS Nano 2017, 12, 24–43. [Google Scholar] [CrossRef]
- Cao, L.; Zhu, Y.; Wang, W.; Wang, G.; Zhang, S.; Cheng, H. Emerging Nano-Based Strategies Against Drug Resistance in Tumor Chemotherapy. Front. Bioeng. Biotechnol. 2021, 9, 798882. [Google Scholar] [CrossRef] [PubMed]
- Soe, Z.C.; Kwon, J.B.; Thapa, R.K.; Ou, W.; Nguyen, H.T.; Gautam, M.; Oh, K.T.; Choi, H.-G.; Ku, S.K.; Yong, C.S.; et al. Transferrin-Conjugated Polymeric Nanoparticle for Receptor-Mediated Delivery of Doxorubicin in Doxorubicin-Resistant Breast Cancer Cells. Pharmaceutics 2019, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Chen, L.; Sui, X.; Wu, B.; Zou, S.; Li, A.; Zhang, Y.; Liu, X.; Wang, D.; Cai, S.; et al. Enhanced delivery of sorafenib with anti-GPC3 antibody-conjugated TPGS-b-PCL/Pluronic P123 polymeric nanoparticles for targeted therapy of hepatocellular carcinoma. Mater. Sci. Eng. C 2018, 91, 395–403. [Google Scholar] [CrossRef]
- Vivek, R.; Thangam, R.; NipunBabu, V.; Rejeeth, C.; Sivasubramanian, S.; Gunasekaran, P.; Muthuchelian, K.; Kannan, S. Multifunctional HER2-Antibody Conjugated Polymeric Nanocarrier-Based Drug Delivery System for Multi-Drug-Resistant Breast Cancer Therapy. ACS Appl. Mater. Interfaces 2014, 6, 6469–6480. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, M.; Dey, R.; Chen, Y. Nanomaterials for cancer therapy: Current progress and perspectives. J. Hematol. Oncol. 2021, 14, 85. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in nanomedicine: Approved and investigational nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Kim, T.H.; Koh, Y.H.; Kim, B.H.; Kim, M.J.; Lee, J.H.; Park, B.; Park, J.-W. Proton beam radiotherapy vs. radiofrequency ablation for recurrent hepatocellular carcinoma: A randomized phase III trial. J. Hepatol. 2020, 74, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A. Drug-eluting beads transarterial chemoembolization for hepatocellular carcinoma: Current state of the art. World J. Gastroenterol. 2018, 24, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Koulouris, A.; Tsagkaris, C.; Spyrou, V.; Pappa, E.; Troullinou, A.; Nikolaou, M. Hepatocellular Carcinoma: An Overview of the Changing Landscape of Treatment Options. J. Hepatocell. Carcinoma 2021, 8, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsui, J.; Matsushima, T.; Obaishi, H.; Miyazaki, K.; Nakamura, K.; Tohyama, O.; Semba, T.; Yamaguchi, A.; Hoshi, S.S.; et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc. Cell 2014, 6, 18. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Shah, N.J.; Kelly, W.J.; Liu, S.V.; Choquette, K.; Spira, A. Product review on the Anti-PD-L1 antibody atezolizumab. Hum. Vaccines Immunother. 2017, 14, 269–276. [Google Scholar] [CrossRef]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2021, 76, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Melero, I.; Yau, T.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Choo, S.; Trojan, J.; Welling, T.; Meyer, T.; et al. Impact of antitumor activity on survival outcomes, and nonconventional benefit, with nivolumab (NIVO) in patients with advanced hepatocellular carcinoma (aHCC): Subanalyses of CheckMate-040. J. Clin. Oncol. 2018, 36, 475. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Cattan, S.; Edeline, J.; Ogasawara, S.; Palmer, D.H.; Verslype, C.; Zagonel, V.; Rosmorduc, O.; Vogel, A.; et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J. Clin. Oncol. 2018, 36 (Suppl. S4), 209. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib Suppresses Tumor Growth and Metastasis in Hepatocellular Carcinoma by a Dual Blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Tsang, J.; Wong, J.S.L.; Kwok, G.G.W.; Li, B.C.W.; Leung, R.; Chiu, J.; Cheung, T.T.; Yau, T. Nivolumab + Ipilimumab for patients with hepatocellular carcinoma previously treated with Sorafenib. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 589–598. [Google Scholar] [CrossRef]
- FDAnews. FDA Grants Priority Review to AstraZeneca’s Adjunct HCC Drug. 25 April 2022. Available online: https://www.fdanews.com/articles/207545-fda-grants-priority-review-to-astrazenecas-adjunct-hcc-drug (accessed on 29 August 2022).
- AstraZeneca. Tremelimumab US Priority Review for Imfinzi Combo. Available online: https://www.astrazeneca.com/media-centre/press-releases/2022/tremelimumab-us-priority-review-imfinzi-combo.html (accessed on 29 August 2022).
- Kudo, M. Durvalumab plus tremelimumab in unresectable hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2022, 11, 592–596. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Gagliardi, A.; Giuliano, E.; Venkateswararao, E.; Fresta, M.; Bulotta, S.; Awasthi, V.; Cosco, D. Biodegradable Polymeric Nanoparticles for Drug Delivery to Solid Tumors. Front. Pharmacol. 2021, 12, 601626. [Google Scholar] [CrossRef] [PubMed]
- Mossenta, M.; Busato, D.; Bo, M.D.; Toffoli, G. Glucose Metabolism and Oxidative Stress in Hepatocellular Carcinoma: Role and Possible Implications in Novel Therapeutic Strategies. Cancers 2020, 12, 1668. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tan, S.; Li, S.; Shen, Q.; Wang, K. Cancer drug delivery in the nano era: An overview and perspectives (Review). Oncol. Rep. 2017, 38, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Ding, T.; Guo, Z.-W.; Yu, X.-J.; Hu, Y.-Z.; Zheng, L.; Xu, J. Expression pattern of tumour-associated antigens in hepatocellular carcinoma: Association with immune infiltration and disease progression. Br. J. Cancer 2013, 109, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Bakrania, A.; Zheng, G.; Bhat, M. Nanomedicine in Hepatocellular Carcinoma: A New Frontier in Targeted Cancer Treatment. Pharmaceutics 2021, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Y.; Ge, L.; Lin, Y.; Kwok, H.F. The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma. Cancers 2018, 10, 82. [Google Scholar] [CrossRef]
- Johnson, P.; Zhou, Q.; Dao, D.Y.; Lo, Y.M.D. Circulating biomarkers in the diagnosis and management of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2022, 1–12. [Google Scholar] [CrossRef]
- Wang, M.; Li, J.; Wang, L.; Chen, X.; Zhang, Z.; Yue, D.; Ping, Y.; Shi, X.; Huang, L.; Zhang, T.; et al. Combined cancer testis antigens enhanced prediction accuracy for prognosis of patients with hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 3513–3528. [Google Scholar]
- Chen, C.-H.; Chen, G.-J.; Lee, H.-S.; Huang, G.-T.; Yang, P.-M.; Tsai, L.-J.; Chen, D.-S.; Sheu, J.-C. Expressions of cancer-testis antigens in human hepatocellular carcinomas. Cancer Lett. 2001, 164, 189–195. [Google Scholar] [CrossRef]
- Bricard, G.; Bouzourene, H.; Martinet, O.; Rimoldi, D.; Halkic, N.; Gillet, M.; Chaubert, P.; MacDonald, H.R.; Romero, P.; Cerottini, J.-C.; et al. Naturally Acquired MAGE-A10- and SSX-2-Specific CD8+ T Cell Responses in Patients with Hepatocellular Carcinoma. J. Immunol. 2005, 174, 1709–1716. [Google Scholar] [CrossRef]
- Tahara, K.; Mori, M.; Sadanaga, N.; Sakamoto, Y.; Kitano, S.; Makuuchi, M. Expression of the MAGE gene family in human hepatocellular carcinoma. Cancer 1999, 85, 1234–1240. [Google Scholar] [CrossRef]
- Kerzerho, J.; Adotevi, O.; Castelli, F.A.; Dosset, M.; Bernardeau, K.; Szely, N.; Lang, F.; Tartour, E.; Maillere, B. The Angiogenic Growth Factor and Biomarker Midkine Is a Tumor-Shared Antigen. J. Immunol. 2010, 185, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.-L.; Ye, Q.-H.; Qin, L.-X.; Budhu, A.; Forgues, M.; Chen, Y.; Liu, Y.-K.; Sun, H.-C.; Wang, L.; Lu, H.-Z.; et al. Gene Expression Profiling Reveals Potential Biomarkers of Human Hepatocellular Carcinoma. Clin. Cancer Res. 2007, 13, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.-X.; et al. EpCAM and α-Fetoprotein Expression Defines Novel Prognostic Subtypes of Hepatocellular Carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Shi, B.; Abrams, M.; Sepp-Lorenzino, L. Expression of Asialoglycoprotein Receptor 1 in Human Hepatocellular Carcinoma. J. Histochem. Cytochem. 2013, 61, 901–909. [Google Scholar] [CrossRef]
- Xiao, C.; Fu, X.; Wang, Y.; Liu, H.; Jiang, Y.; Zhao, Z.; You, F. Transferrin receptor regulates malignancies and the stemness of hepatocellular carcinoma-derived cancer stem-like cells by affecting iron accumulation. PLoS ONE 2020, 15, e0243812. [Google Scholar] [CrossRef]
- Adachi, M.; Kai, K.; Yamaji, K.; Ide, T.; Noshiro, H.; Kawaguchi, A.; Aishima, S. Transferrin receptor 1 overexpression is associated with tumour de-differentiation and acts as a potential prognostic indicator of hepatocellular carcinoma. Histopathology 2019, 75, 63–73. [Google Scholar] [CrossRef]
- Fernández, M.; Javaid, F.; Chudasama, V. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2018, 9, 790–810. [Google Scholar] [CrossRef]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef]
- Jung, S.-M.; Tsai, F.-C.; Shiu, T.-F.; Wu, H.-H.; Lin, Y.-S.; Yeh, C.-N.; Chen, Y.-C.; Chu, P.-H. MUC1, MUC2 and MUC5AC expression in hepatocellular carcinoma with cardiac metastasis. Mol. Med. Rep. 2009, 2, 291–294. [Google Scholar] [CrossRef]
- Yuan, S.-F.; Li, K.-Z.; Wang, L.; Dou, K.-F.; Yan, Z.; Han, W.; Zhang, Y.-Q. Expression of MUC1 and its significance in hepatocellular and cholangiocarcinoma tissue. World J. Gastroenterol. 2005, 11, 4661–4666. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Morine, Y.; Ikemoto, T.; Imura, S.; Higashijima, J.; Iwahashi, S.; Saito, Y.; Takasu, C.; Yamada, S.; Ishikawa, D.; et al. Elevated Preoperative Serum CEA Level Is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma through the Epithelial–Mesenchymal Transition. Anticancer Res. 2017, 37, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, S.-W. The Roles of Carcinoembryonic Antigen in Liver Metastasis and Therapeutic Approaches. Gastroenterol. Res. Pract. 2017, 2017, 7521987. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Li, Y.; Yang, F.; Han, D.; Wu, J.; Shi, S.; Tian, F.; Guo, Z.; Xi, W.; Li, G.; et al. Expression of Prostate-Specific Membrane Antigen in Tumor-Associated Vasculature Predicts Poor Prognosis in Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00041-7. [Google Scholar] [CrossRef]
- Christian, S.; Ahorn, H.; Koehler, A.; Eisenhaber, F.; Rodi, H.-P.; Garin-Chesa, P.; Park, J.E.; Rettig, W.J.; Lenter, M.C. Molecular Cloning and Characterization of Endosialin, a C-type Lectin-like Cell Surface Receptor of Tumor Endothelium. J. Biol. Chem. 2001, 276, 7408–7414. [Google Scholar] [CrossRef]
- Christian, S.; Winkler, R.; Helfrich, I.; Boos, A.M.; Besemfelder, E.; Schadendorf, D.; Augustin, H.G. Endosialin (Tem1) Is a Marker of Tumor-Associated Myofibroblasts and Tumor Vessel-Associated Mural Cells. Am. J. Pathol. 2008, 172, 486–494. [Google Scholar] [CrossRef]
- Mogler, C.; König, C.; Wieland, M.; Runge, A.; Besemfelder, E.; Komljenovic, D.; Longerich, T.; Schirmacher, P.; Augustin, H.G. Hepatic stellate cells limit hepatocellular carcinoma progression through the orphan receptor endosialin. EMBO Mol. Med. 2017, 9, 741–749. [Google Scholar] [CrossRef]
- Shimizu, Y.; Suzuki, T.; Yoshikawa, T.; Endo, I.; Nakatsura, T. Next-Generation Cancer Immunotherapy Targeting Glypican-3. Front. Oncol. 2019, 9, 248. [Google Scholar] [CrossRef]
- Xiao, Y.; Yan, W.; Guo, L.; Meng, C.; Li, B.; Neves, H.; Chen, P.-C.; Li, L.; Huang, Y.; Kwok, H.F.; et al. Digitoxin synergizes with sorafenib to inhibit hepatocelluar carcinoma cell growth without inhibiting cell migration. Mol. Med. Rep. 2016, 15, 941–947. [Google Scholar] [CrossRef]
- Xiao, W.; Zhao, S.; Shen, F.; Liang, J.; Chen, J. Overexpression of CD147 is associated with poor prognosis, tumor cell migration and ERK signaling pathway activation in hepatocellular carcinoma. Exp. Ther. Med. 2017, 14, 2637–2642. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Long, X.; Zhang, J.; Huang, X.; Aa, J.; Yang, H.; Chen, Z.; Xing, J. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARα pathways. J. Hepatol. 2015, 63, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, J.; Xing, J.; Li, W.; Li, H.; Ke, X.; Zhang, J.; Ren, T.; Shang, Y.; Yang, H.; et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J. Hepatol. 2014, 61, 859–866. [Google Scholar] [CrossRef]
- Ito, H.; Funahashi, S.-I.; Yamauchi, N.; Shibahara, J.; Midorikawa, Y.; Kawai, S.; Kinoshita, Y.; Watanabe, A.; Hippo, Y.; Ohtomo, T.; et al. Identification of ROBO1 as a Novel Hepatocellular Carcinoma Antigen and a Potential Therapeutic and Diagnostic Target. Clin. Cancer Res. 2006, 12, 3257–3264. [Google Scholar] [CrossRef]
- Ao, J.-Y.; Chai, Z.-T.; Zhang, Y.-Y.; Zhu, X.-D.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Cai, H.; Gao, D.-M.; Sun, H.-C. Robo1 promotes angiogenesis in hepatocellular carcinoma through the Rho family of guanosine triphosphatases’ signaling pathway. Tumor Biol. 2015, 36, 8413–8424. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Z. Immunostaining of PD-1/PD-Ls in liver tissues of patients with hepatitis and hepatocellular carcinoma. World J. Gastroenterol. 2011, 17, 3322–3329. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-L.; Liu, L.-P.; Jiang, J.-X.; Xiong, Z.-F.; He, Q.-J.; Wu, C. The Correlation of Expression Levels of HIF-1 and HIF-2 in Hepatocellular Carcinoma with Capsular Invasion, Portal Vein Tumor Thrombi and Patients’ Clinical Outcome. Jpn. J. Clin. Oncol. 2013, 44, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zhang, H.; Zheng, J.; Liu, Y. Glypican-3: A New Target for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Cancer 2020, 11, 2008–2021. [Google Scholar] [CrossRef]
- Nishida, T.; Kataoka, H. Glypican 3-Targeted Therapy in Hepatocellular Carcinoma. Cancers 2019, 11, 1339. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, X.; Lei, Y.; Wang, G.; Liu, M. Glypican-3: A Novel and Promising Target for the Treatment of Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 824208. [Google Scholar] [CrossRef]
- Filmus, J.; Capurro, M. The role of glypican-3 in the regulation of body size and cancer. Cell Cycle 2008, 7, 2787–2790. [Google Scholar] [CrossRef]
- Veugelers, M.; De Cat, B.; Ceulemans, H.; Bruystens, A.-M.; Coomans, C.; Dürr, J.; Vermeesch, J.; Marynen, P.; David, G. Glypican-6, a New Member of the Glypican Family of Cell Surface Heparan Sulfate Proteoglycans. J. Biol. Chem. 1999, 274, 26968–26977. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Church, J.G.; Buick, R.N. Isolation of a cDNA corresponding to a developmentally regulated transcript in rat intestine. Mol. Cell. Biol. 1988, 8, 4243–4249. [Google Scholar] [CrossRef] [PubMed]
- Pilia, G.; Hughes-Benzie, R.M.; MacKenzie, A.; Baybayan, P.; Chen, E.Y.; Huber, R.; Neri, G.; Cao, A.; Forabosco, A.; Schlessinger, D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet. 1996, 12, 241–247. [Google Scholar] [CrossRef] [PubMed]
- De Cat, B.; David, G. Developmental roles of the glypicans. Semin. Cell Dev. Biol. 2001, 12, 117–125. [Google Scholar] [CrossRef]
- Hippo, Y.; Watanabe, K.; Watanabe, A.; Midorikawa, Y.; Yamamoto, S.; Ihara, S.; Tokita, S.; Iwanari, H.; Ito, Y.; Nakano, K.; et al. Identification of Soluble NH2-Terminal Fragment of Glypican-3 as a Serological Marker for Early-Stage Hepatocellular Carcinoma. Cancer Res. 2004, 64, 2418–2423. [Google Scholar] [CrossRef]
- De Cat, B.; Muyldermans, S.-Y.; Coomans, C.; DeGeest, G.; Vanderschueren, B.; Creemers, J.; Biemar, F.; Peers, B.; David, G. Processing by proprotein convertases is required for glypican-3 modulation of cell survival, Wnt signaling, and gastrulation movements. J. Cell Biol. 2003, 163, 625–635. [Google Scholar] [CrossRef]
- Ho, M.; Kim, H. Glypican-3: A new target for cancer immunotherapy. Eur. J. Cancer 2011, 47, 333–338. [Google Scholar] [CrossRef]
- Filmus, J. Glypicans in growth control and cancer. Glycobiology 2001, 11, 19–23. [Google Scholar] [CrossRef]
- Montalbano, M.; Curcurù, G.; Shirafkan, A.; Vento, R.; Rastellini, C.; Cicalese, L. Modeling of Hepatocytes Proliferation Isolated from Proximal and Distal Zones from Human Hepatocellular Carcinoma Lesion. PLoS ONE 2016, 11, e0153613. [Google Scholar] [CrossRef]
- Capurro, M.I.; Xiang, Y.-Y.; Lobe, C.; Filmus, J. Glypican-3 Promotes the Growth of Hepatocellular Carcinoma by Stimulating Canonical Wnt Signaling. Cancer Res. 2005, 65, 6245–6254. [Google Scholar] [CrossRef]
- Capurro, M.; Martin, T.; Shi, W.; Filmus, J. Glypican-3 binds to frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J. Cell Sci. 2014, 127, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Song, H.H.; Shi, W.; Xiang, Y.-Y.; Filmus, J. The Loss of Glypican-3 Induces Alterations in Wnt Signaling. J. Biol. Chem. 2005, 280, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Tomar, P.C. Hybridoma technology; advancements, clinical significance, and future aspects. J. Genet. Eng. Biotechnol. 2021, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Alibakhshi, A.; Kahaki, F.A.; Ahangarzadeh, S.; Yaghoobi, H.; Yarian, F.; Arezumand, R.; Ranjbari, J.; Mokhtarzadeh, A.; de la Guardia, M. Targeted cancer therapy through antibody fragments-decorated nanomedicines. J. Control. Release 2017, 268, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef]
- Ishiguro, T.; Sugimoto, M.; Kinoshita, Y.; Miyazaki, Y.; Nakano, K.; Tsunoda, H.; Sugo, I.; Ohizumi, I.; Aburatani, H.; Hamakubo, T.; et al. Anti–Glypican 3 Antibody as a Potential Antitumor Agent for Human Liver Cancer. Cancer Res. 2008, 68, 9832–9838. [Google Scholar] [CrossRef]
- Zhu, A.X.; Gold, P.J.; El-Khoueiry, A.B.; Abrams, T.A.; Morikawa, H.; Ohishi, N.; Ohtomo, T.; Philip, P.A. First-in-Man Phase I Study of GC33, a Novel Recombinant Humanized Antibody against Glypican-3, in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2013, 19, 920–928. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Puig, O.; Daniele, B.; Kudo, M.; Merle, P.; Park, J.-W.; Ross, P.; Peron, J.-M.; Ebert, O.; Chan, S.; et al. Randomized phase II placebo controlled study of codrituzumab in previously treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2016, 65, 289–295. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Yen, C.-J.; Hsu, C.-H.; O’Donoghue, J.; Beylergil, V.; Ruan, S.; Pandit-Taskar, N.; Gansukh, B.; Lyashchenko, S.K.; Ma, J.; et al. Phase Ib study of codrituzumab in combination with sorafenib in patients with non-curable advanced hepatocellular carcinoma (HCC). Cancer Chemother. Pharmacol. 2017, 79, 421–429. [Google Scholar] [CrossRef]
- Phung, Y.; Gao, W.; Man, Y.-G.; Nagata, S.; Ho, M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. mAbs 2012, 4, 592–599. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Ho, M. Humanization of high-affinity antibodies targeting glypican-3 in hepatocellular carcinoma. Sci. Rep. 2016, 6, srep33878. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Zhang, H.; Ye, W.; Huttad, L.; Tan, M.; Chua, M.-S.; Gambhir, S.; So, S. A Humanized Anti-GPC3 Antibody for Immuno-Positron Emission Tomography Imaging of Orthotopic Mouse Model of Patient-Derived Hepatocellular Carcinoma Xenografts. Cancers 2021, 13, 3977. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Gao, W.; Wang, R.; Chen, W.; Man, Y.-G.; Figg, W.D.; Wang, X.W.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, E1083–E1091. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Zhang, D.; Zhang, Y.; Wang, C.; Shi, L.; Wei, W.; Huang, G.; Liu, J. GPC3-targeted immunoPET imaging of hepatocellular carcinomas. Eur. J. Pediatr. 2022, 49, 2682–2692. [Google Scholar] [CrossRef]
- Ishiguro, T.; Sano, Y.; Komatsu, S.-I.; Kamata-Sakurai, M.; Kaneko, A.; Kinoshita, Y.; Shiraiwa, H.; Azuma, Y.; Tsunenari, T.; Kayukawa, Y.; et al. An anti–glypican 3/CD3 bispecific T cell–redirecting antibody for treatment of solid tumors. Sci. Transl. Med. 2017, 9, eaal4291. [Google Scholar] [CrossRef]
- Waaijer, S.J.; Giesen, D.; Ishiguro, T.; Sano, Y.; Sugaya, N.; Schröder, C.P.; de Vries, E.G.; Hooge, M.N.L.-D. Preclinical PET imaging of bispecific antibody ERY974 targeting CD3 and glypican 3 reveals that tumor uptake correlates to T cell infiltrate. J. Immunother. Cancer 2019, 8, e000548. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.Gov. ERY974 and Hepatocellular Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT05022927?term=ery974&cond=hepatocellular+carcinoma&draw=2&rank=1 (accessed on 22 August 2022).
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef]
- Batra, J.K.; Kasprzyk, P.G.; Bird, R.E.; Pastan, I.; King, C.R. Recombinant Anti-ErbB2 Immunotoxins Containing Pseudomonas Exotoxin. Proc. Natl. Acad. Sci. USA 1992, 89, 5867–5871. [Google Scholar] [CrossRef]
- Yeganeh, H.H.; Heiat, M.; Kieliszek, M.; Alavian, S.M.; Rezaie, E. DT389-YP7, a Recombinant Immunotoxin against Glypican-3 That Inhibits Hepatocellular Cancer Cells: An In Vitro Study. Toxins 2021, 13, 749. [Google Scholar] [CrossRef]
- Wang, C.; Gao, W.; Feng, M.; Pastan, I.; Ho, M. Construction of an immunotoxin, HN3-mPE24, targeting glypican-3 for liver cancer therapy. Oncotarget 2016, 8, 32450–32460. [Google Scholar] [CrossRef]
- Fleming, B.D.; Urban, D.J.; Hall, M.D.; Longerich, T.; Greten, T.F.; Pastan, I.; Ho, M. Engineered Anti-GPC3 Immunotoxin, HN3-ABD-T20, Produces Regression in Mouse Liver Cancer Xenografts Through Prolonged Serum Retention. Hepatology 2019, 71, 1696–1711. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2017, 7, 690. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S.; et al. Glypican-3–Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Qian, S.; Huang, M.; Chen, M.; Peng, L.; Liu, J.; Xu, W.; Xu, J. Development of GPC3 and EGFR-dual-targeting chimeric antigen receptor-T cells for adoptive T cell therapy. Am. J. Transl. Res. 2021, 13, 156–167. [Google Scholar] [PubMed]
- ClinicalTrials.Gov. CAR-T and Hepatocellular Carcinoma. Available online: https://clinicaltrials.gov/ct2/results?term=CAR-T&cond=hepatocellular+carcinoma&Search=Apply&recrs=a&recrs=d&recrs=e&age_v=&gndr=&type=&rslt= (accessed on 23 August 2022).
- Fang, W.; Fu, Q.; Zhao, Q.; Zheng, Y.; Liu, L.; Li, Z.; Dai, X.; Wang, H.; Zhu, X.; Zhao, P.; et al. Phase I trial of fourth-generation chimeric antigen receptor T-cells targeting glypican-3 for advanced hepatocellular carcinoma. J. Clin. Oncol. 2021, 39, 4088. [Google Scholar] [CrossRef]
- Zhai, B.; Shi, D.; Gao, H.; Qi, X.; Jiang, H.; Zhang, Y.; Chi, J.; Ruan, H.; Wang, H.; Ru, Q.C.; et al. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC). J. Clin. Oncol. 2017, 35, 3049. [Google Scholar] [CrossRef]
- La Lee, Y.; Ahn, B.-C.; Lee, Y.; Lee, S.-W.; Cho, J.-Y.; Lee, J. Targeting of hepatocellular carcinoma with glypican-3-targeting peptide ligand. J. Pept. Sci. 2011, 17, 763–769. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, J.; Wang, Y.; Wang, G.; Wang, X.; Zhou, Z.; Liu, G.; Gao, S.; Zhu, L. Identification of a Glypican-3-Binding Peptide for In Vivo Non-Invasive Human Hepatocellular Carcinoma Detection. Macromol. Biosci. 2016, 17. [Google Scholar] [CrossRef]
- Zhu, D.; Qin, Y.; Wang, J.; Zhang, L.; Zou, S.; Zhu, X.; Zhu, L. Novel Glypican-3-Binding Peptide for in Vivo Hepatocellular Carcinoma Fluorescent Imaging. Bioconj. Chem. 2016, 27, 831–839. [Google Scholar] [CrossRef]
- Berman, R.M.; Kelada, O.J.; Gutsche, N.T.; Natarajan, R.; Swenson, R.E.; Fu, Y.; Hong, J.; Ho, M.; Choyke, P.L.; Escorcia, F.E. In Vitro Performance of Published Glypican 3-Targeting Peptides TJ12P1 and L5 Indicates Lack of Specificity and Potency. Cancer Biother. Radiopharm. 2019, 34, 498–503. [Google Scholar] [CrossRef]
- Sawada, Y.; Sakai, M.; Yoshikawa, T.; Ofuji, K.; Nakatsura, T. A glypican-3-derived peptide vaccine against hepatocellular carcinoma. OncoImmunology 2012, 1, 1448–1450. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Yoshikawa, T.; Nobuoka, D.; Shirakawa, H.; Kuronuma, T.; Motomura, Y.; Mizuno, S.; Ishii, H.; Nakachi, K.; Konishi, M.; et al. Phase I Trial of a Glypican-3–Derived Peptide Vaccine for Advanced Hepatocellular Carcinoma: Immunologic Evidence and Potential for Improving Overall Survival. Clin. Cancer Res. 2012, 18, 3686–3696. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Yoshikawa, T.; Ofuji, K.; Yoshimura, M.; Tsuchiya, N.; Takahashi, M.; Nobuoka, D.; Gotohda, N.; Takahashi, S.; Kato, Y.; et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. OncoImmunology 2016, 5, e1129483. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Okusaka, T.; Ohno, I.; Mitsunaga, S.; Kondo, S.; Ueno, H.; Morizane, C.; Gemmoto, K.; Suna, H.; Ushida, Y.; et al. Phase I studies of peptide vaccine cocktails derived from GPC3, WDRPUH and NEIL3 for advanced hepatocellular carcinoma. Immunotherapy 2021, 13, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.M.; Gutsche, N.T.; King, A.P.; Baidoo, K.E.; Kelada, O.J.; Choyke, P.L.; Escorcia, F.E. Glypican-3-Targeted Alpha Particle Therapy for Hepatocellular Carcinoma. Molecules 2020, 26, 4. [Google Scholar] [CrossRef]
- Labadie, K.P.; Hamlin, D.K.; Kenoyer, A.; Daniel, S.K.; Utria, A.F.; Ludwig, A.D.; Kenerson, H.L.; Li, L.; Sham, J.G.; Chen, D.L.; et al. Glypican-3–Targeted 227Th α-Therapy Reduces Tumor Burden in an Orthotopic Xenograft Murine Model of Hepatocellular Carcinoma. J. Nucl. Med. 2021, 63, 1033–1038. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Lei, C.; Liu, X.-R.; Chen, Q.-B.; Li, Y.; Zhou, J.-L.; Zhou, L.-Y.; Zou, T. Hyaluronic acid and albumin based nanoparticles for drug delivery. J. Control. Release 2021, 331, 416–433. [Google Scholar] [CrossRef] [PubMed]
- Hornok, V. Serum Albumin Nanoparticles: Problems and Prospects. Polymers 2021, 13, 3759. [Google Scholar] [CrossRef]
- Rizeq, B.R.; Younes, N.N.; Rasool, K.; Nasrallah, G.K. Synthesis, Bioapplications, and Toxicity Evaluation of Chitosan-Based Nanoparticles. Int. J. Mol. Sci. 2019, 20, 5776. [Google Scholar] [CrossRef]
- Aibani, N.; Rai, R.; Patel, P.; Cuddihy, G.; Wasan, E.K. Chitosan Nanoparticles at the Biological Interface: Implications for Drug Delivery. Pharmaceutics 2021, 13, 1686. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lillard, J.W., Jr. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.K.; Kokaz, S.F.; Abed, S.N.; Paradkar, A.; Tekade, R.K. Chapter 7—Levels of drug Targeting. In Basic Fundamentals of Drug Delivery; Tekade, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 203–267. ISBN 9780128179093. [Google Scholar]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Banik, B.L.; Fattahi, P.; Brown, J.L. Polymeric nanoparticles: The future of nanomedicine. WIREs Nanomed. Nanobiotechnol. 2015, 8, 271–299. [Google Scholar] [CrossRef]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surfaces B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Das, S.S.; Bharadwaj, P.; Bilal, M.; Barani, M.; Rahdar, A.; Taboada, P.; Bungau, S.; Kyzas, G.Z. Stimuli-Responsive Polymeric Nanocarriers for Drug Delivery, Imaging, and Theragnosis. Polymers 2020, 12, 1397. [Google Scholar] [CrossRef]
- AlSawaftah, N.M.; Awad, N.S.; Pitt, W.G.; Husseini, G.A. pH-Responsive Nanocarriers in Cancer Therapy. Polymers 2022, 14, 936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Shi, Z.; Yang, Y.; Xie, X.; Lee, S.M.; Wang, Y.; Leong, K.W.; Chen, M. pH-sensitive polymeric nanoparticles for co-delivery of doxorubicin and curcumin to treat cancer via enhanced pro-apoptotic and anti-angiogenic activities. Acta Biomater. 2017, 58, 349–364. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Yu, L.; Yang, Y.; Zou, B.; Ma, W.; Yu, M.; Lu, J.; Xiong, G.; Yu, Z.; Li, A. Delivery of triptolide with reduction-sensitive polymer nanoparticles for liver cancer therapy on patient-derived xenografts models. Chin. Chem. Lett. 2020, 31, 3178–3182. [Google Scholar] [CrossRef]
- Lu, C.-T.; Zhao, Y.-Z.; Ge, S.-P.; Jin, Y.-G.; Du, L.-N. Potential and problems in ultrasound-responsive drug delivery systems. Int. J. Nanomed. 2013, 8, 1621–1633. [Google Scholar] [CrossRef] [Green Version]
- Marano, F.; Argenziano, M.; Frairia, R.; Adamini, A.; Bosco, O.; Rinella, L.; Fortunati, N.; Cavalli, R.; Catalano, M.G. Doxorubicin-Loaded Nanobubbles Combined with Extracorporeal Shock Waves: Basis for a New Drug Delivery Tool in Anaplastic Thyroid Cancer. Thyroid 2016, 26, 705–716. [Google Scholar] [CrossRef]
- Du, L.; Jin, Y.; Zhou, W.; Zhao, J. Ultrasound-Triggered Drug Release and Enhanced Anticancer Effect of Doxorubicin-Loaded Poly(D,L-Lactide-Co-Glycolide)-Methoxy-Poly(Ethylene Glycol) Nanodroplets. Ultrasound Med. Biol. 2011, 37, 1252–1258. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; He, C.; Jing, Z.; Liu, C.; Xie, J.; Ma, W.; Deng, H. The use of 5-fluorouracil-loaded nanobubbles combined with low-frequency ultrasound to treat hepatocellular carcinoma in nude mice. Eur. J. Med. Res. 2017, 22, 48. [Google Scholar] [CrossRef]
- Tang, X.; Chen, L.; Li, A.; Cai, S.; Zhang, Y.; Liu, X.; Jiang, Z.; Liu, X.; Liang, Y.; Ma, D. Anti-GPC3 antibody-modified sorafenib-loaded nanoparticles significantly inhibited HepG2 hepatocellular carcinoma. Drug Deliv. 2018, 25, 1484–1494. [Google Scholar] [CrossRef]
- Shen, J.; Cai, W.; Ma, Y.; Xu, R.; Huo, Z.; Song, L.; Qiu, X.; Zhang, Y.; Li, A.; Cao, W.; et al. hGC33-Modified and Sorafenib-Loaded Nanoparticles have a Synergistic Anti-Hepatoma Effect by Inhibiting Wnt Signaling Pathway. Nanoscale Res. Lett. 2020, 15, 220. [Google Scholar] [CrossRef]
- Mu, W.; Jiang, D.; Mu, S.; Liang, S.; Liu, Y.; Zhang, N. Promoting Early Diagnosis and Precise Therapy of Hepatocellular Carcinoma by Glypican-3-Targeted Synergistic Chemo-Photothermal Theranostics. ACS Appl. Mater. Interfaces 2019, 11, 23591–23604. [Google Scholar] [CrossRef]
- Huang, Z.; Li, F.; Zhang, J.; Shi, X.; Xu, Y.; Huang, X. Research on the Construction of Bispecific-Targeted Sustained-Release Drug-Delivery Microspheres and Their Function in Treatment of Hepatocellular Carcinoma. ACS Omega 2022, 7, 22003–22014. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Chu, Q.; Yang, H.; Guan, L.; Fu, S.; Gao, T.; Sang, X.; Zhang, Z.; Liang, S.; Liu, Y.; et al. Multipoint Costriking Nanodevice Eliminates Primary Tumor Cells and Associated-Circulating Tumor Cells for Enhancing Metastasis Inhibition and Therapeutic Effect on HCC. Adv. Sci. 2022, 9, 2101472. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xiao, X.; Li, X.; Xu, Y.; Ma, L.; Guo, L.; Yan, C.; Wu, Y. Detecting GPC3-Expressing Hepatocellular Carcinoma with L5 Peptide-Guided Pretargeting Approach: In Vitro and In Vivo MR Imaging Experiments. Contrast Media Mol. Imaging 2018, 2018, 9169072. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Y.; Liu, X.; Tian, Y.; Cheng, Y.; Tang, L.; Lin, H. Smart NIR-II croconaine dye-peptide for enhanced photo-sonotheranostics of hepatocellular carcinoma. Theranostics 2022, 12, 76–86. [Google Scholar] [CrossRef]
- Di Paola, M.; Quarta, A.; Conversano, F.; Sbenaglia, E.A.; Bettini, S.; Valli, L.; Gigli, G.; Casciaro, S. Human Hepatocarcinoma Cell Targeting by Glypican-3 Ligand Peptide Functionalized Silica Nanoparticles: Implications for Ultrasound Molecular Imaging. Langmuir 2017, 33, 4490–4499. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Molecule Type | Line Treatment and Year of Approval |
|---|---|---|
| Sorafenib | Multi-tyrosine kinase inhibitor | First line, standard of care until 2020 Approved in 2008 [66,67] |
| Regorafenib | Multi-tyrosine kinase inhibitor | Second line Approved in 2017 [66,68] |
| Lenvatinib | Multikinase inhibitor | First line Approved in 2018 [66,69,70] |
| Atezolizumab + bevacizumab | Anti-PD-L1 antibody + anti-VEGF antibody | First line, standard of care Approved in 2020 [66,71,72,73,74] |
| Nivolumab | Anti-PD1 antibody | Second line Approved in 2017 [66,75,76] |
| Pembrolizumab | Anti-PD1 antibody | Second line Approved in 2018 [66,77] |
| Cabozantinib | Tyrosine kinase inhibitor | Second line Approved in 2019 [66,78,79] |
| Ramucirumab | Anti-VEGFR-2 antibody | Second line Approved in 2019 [66,80] |
| Nivolumab + ipilimumab | Anti-PD1 antibody + anti-CTLA-4 antibody | Second line Approved in 2020 [66,81] |
| Durvalumab + Tremelimumab | Anti-PD-L1 + Anti-CTLA-4 | FDA grants priority review to AstraZeneca’s Biologics License Application April 2022 First line [82,83,84,85] |
| Durvalumab | Anti-PD-L1 | Supplemental Biologics License Application has been submitted to FDA April 2022 First line [82,83,84,85] |
| Matrix Component | Features | Advantages | Disadvantages |
|---|---|---|---|
| poly(lactic acid) (PLA), poly(lactic-co-glycolic acid) (PLGA) | Synthetic origin. Biocompatible. Biodegradable. Non-toxic. Negatively charged. [177] | Synthesis can be done with different molecular weights and lactic:glycolic acid ratios. Different NPs shapes can be made. Sustained release of loaded drugs. Surface modifications are possible. PLA-based NPs have been proposed to increase the oral bioavailability of poorly water-soluble drugs. [177] | Poor drug loading. Some formulations have an initial high burst drug release. [177] |
| Albumin (AL) | Natural origin. Biocompatible. Biodegradable. Non-immunogenic. Water-soluble. Negatively charged. [178,179] | Rich in functional groups for ligand/drug binding. AL-based NPs easily cleared in vivo. It is naturally internalized in tumor stroma through the gp60 pathway. High levels of albumin can be supplemented into the body without or with low effects. [178,179] | Some formulations need toxic cross-linking with drugs to increase NPs stability and avoid a burst release. [179] |
| Chitosan (CS) | Natural origin. Biocompatible. Biodegradable. Non-toxic. Positively charged. [180] | It presents hydroxyl and amine functional groups for the addition of crosslinking agents. Positive charge allows CS to attach to cells, tending to accumulate in negatively charged cancer cells, and increase cellular uptake. pH sensibility prevents drug release at physiologic pH (~7.4) and increases it in the acid tumor environment. [180,181] | Low solubility at physiologic pH. Fast dissolution in stomach. [180] |
| Cholesterol and lipid layer (Liposomes) | Natural origin. Non-toxic. Biocompatible. Biodegradable. Non-immunogenic. Sphere-shaped. [176] | Modification of the lipid layer structure to imitate biophysical characteristics of cells. Reduce toxic drug exposures of sensitive tissues. [176] | Low solubility. Short half-life. Possible leakage and fusion of drug/molecules encapsulated. [176] |
| Stimulus | NPs Type | Properties | In HCC |
|---|---|---|---|
| pH | Anionic: Poly(aspartic acid) Poly(acrylic acid) Poly (methacrylic acid) Poly-sulfonamides Cationic: Poly(b-amino ester) Poly(N,N-dimethylamino ethyl methacrylate) poly(L-histidine) [189] | Ionizable groups in NPs structure. Imbalance in hydrophilic-hydrophobic equilibrium at low pH. Disruption of NPs structure with the consequent drug release. [189] | poly(b-amino ester) copolymer NPs loaded with doxorubicin and curcumin had an enhanced and rapid release in HCC acidic environment (pH 5.8) and an increase in cellular uptake. In vivo NPs showed an increased tumor weight inhibition compared to free drugs [190] |
| Redox | Disulfide-linked [188] | Exploit the high levels of glutathione in cancer tissues. Presence of disulfide bonds, which are disrupted in presence of glutathione. [188] | NPs made of a polymer with isocyanate and bis(2-hydroxyethyl)-disulfide with the terminal end pegylated. Triptolide were loaded into NPs. Triptolide is rapidly release in reductive conditions with improved antitumor efficacy and low toxicity. [191] |
| Ultrasound (US) | Nanobubbles (NBs) [192,193] | External biodegradable shell. Inner core of vaporizable compound or gas (perfluorocarbons or sulfur hexafluoride). US delivery based on bubble cavitation and increased cell permeability. [192,193] | Lina Du and colleagues investigated the effect of doxorubicin-loaded PLGA-mPEG NBs in the treatment of mice with subcutaneous H22 tumor (mouse hepatocellular carcinoma cell line). Doxorubicin-NBs showed a reduction in tumor growth compared to mice treated with saline. This effect was further enhanced by the additional external stimulus of US. Lipidic-shelled NBs loaded with 5-fluorouracil showed antitumor effect in HepG2 tumor-bearing mice, with a greater extent once US were administered. [194,195] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mossenta, M.; Busato, D.; Dal Bo, M.; Macor, P.; Toffoli, G. Novel Nanotechnology Approaches to Overcome Drug Resistance in the Treatment of Hepatocellular Carcinoma: Glypican 3 as a Useful Target for Innovative Therapies. Int. J. Mol. Sci. 2022, 23, 10038. https://doi.org/10.3390/ijms231710038
Mossenta M, Busato D, Dal Bo M, Macor P, Toffoli G. Novel Nanotechnology Approaches to Overcome Drug Resistance in the Treatment of Hepatocellular Carcinoma: Glypican 3 as a Useful Target for Innovative Therapies. International Journal of Molecular Sciences. 2022; 23(17):10038. https://doi.org/10.3390/ijms231710038
Chicago/Turabian StyleMossenta, Monica, Davide Busato, Michele Dal Bo, Paolo Macor, and Giuseppe Toffoli. 2022. "Novel Nanotechnology Approaches to Overcome Drug Resistance in the Treatment of Hepatocellular Carcinoma: Glypican 3 as a Useful Target for Innovative Therapies" International Journal of Molecular Sciences 23, no. 17: 10038. https://doi.org/10.3390/ijms231710038