
Synthesis and NLRP3-Inflammasome Inhibitory Activity of the Naturally Occurring Velutone F and of Its Non-Natural Regioisomeric Chalconoids

 ,
,  , , , , , , , , , , and
, , , , , , , , , , and 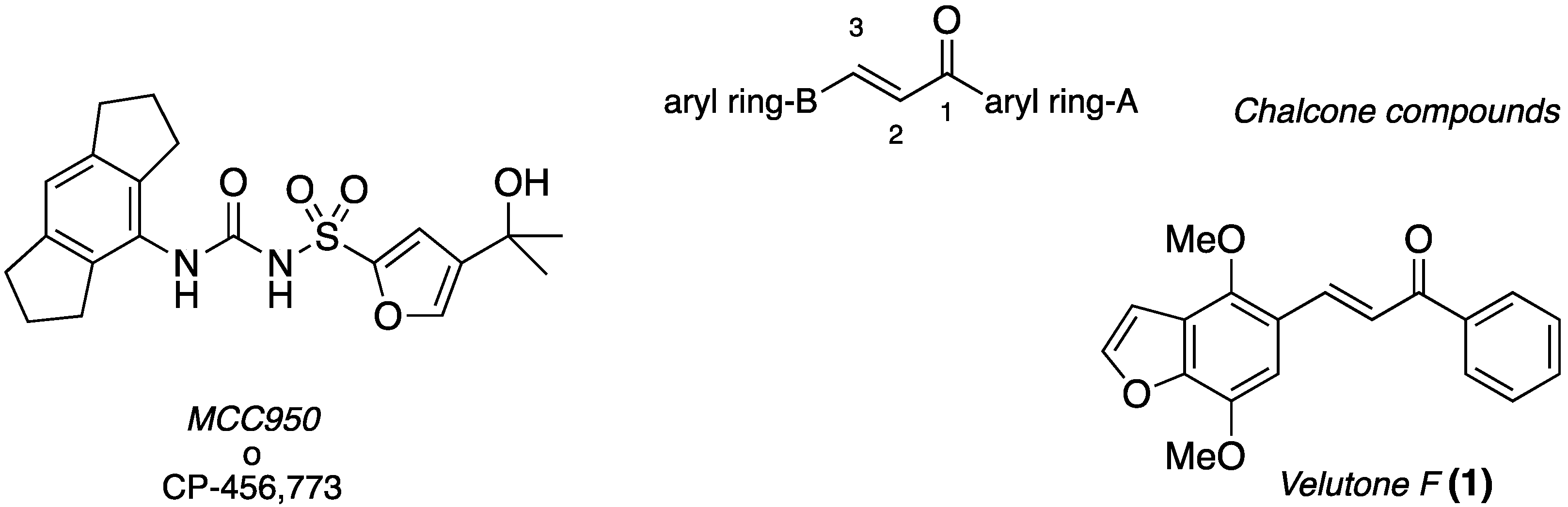{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
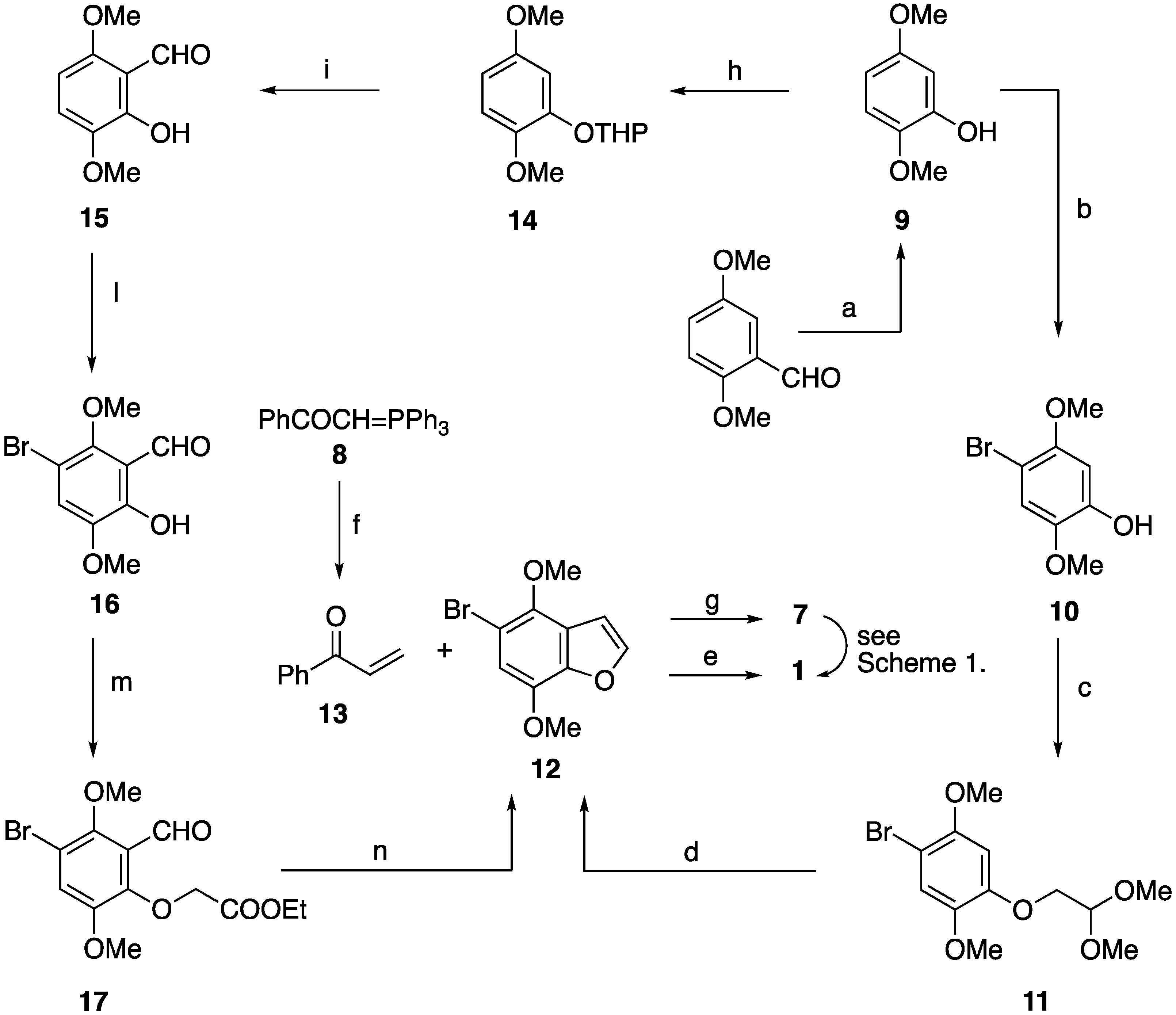
2.1. Synthetic Pathway A for Target Compound 1
2.2. Synthetic Pathway B for Target Compound 1
2.3. Synthetic Pathways Providing the Non-Natural Regioisomers of Velutone F
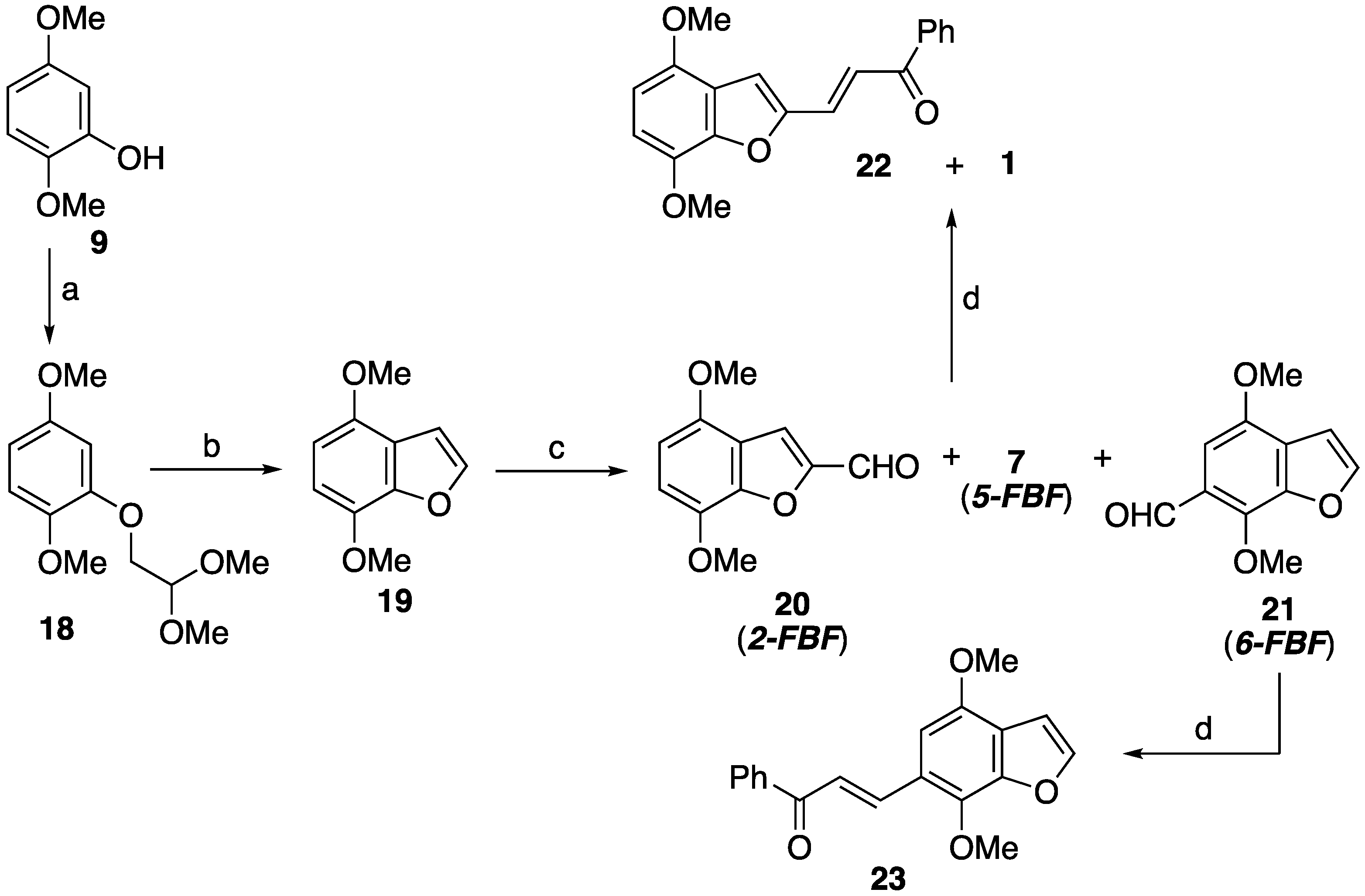
2.3.1. Synthetic Pathways to the Isomers 22 and 23
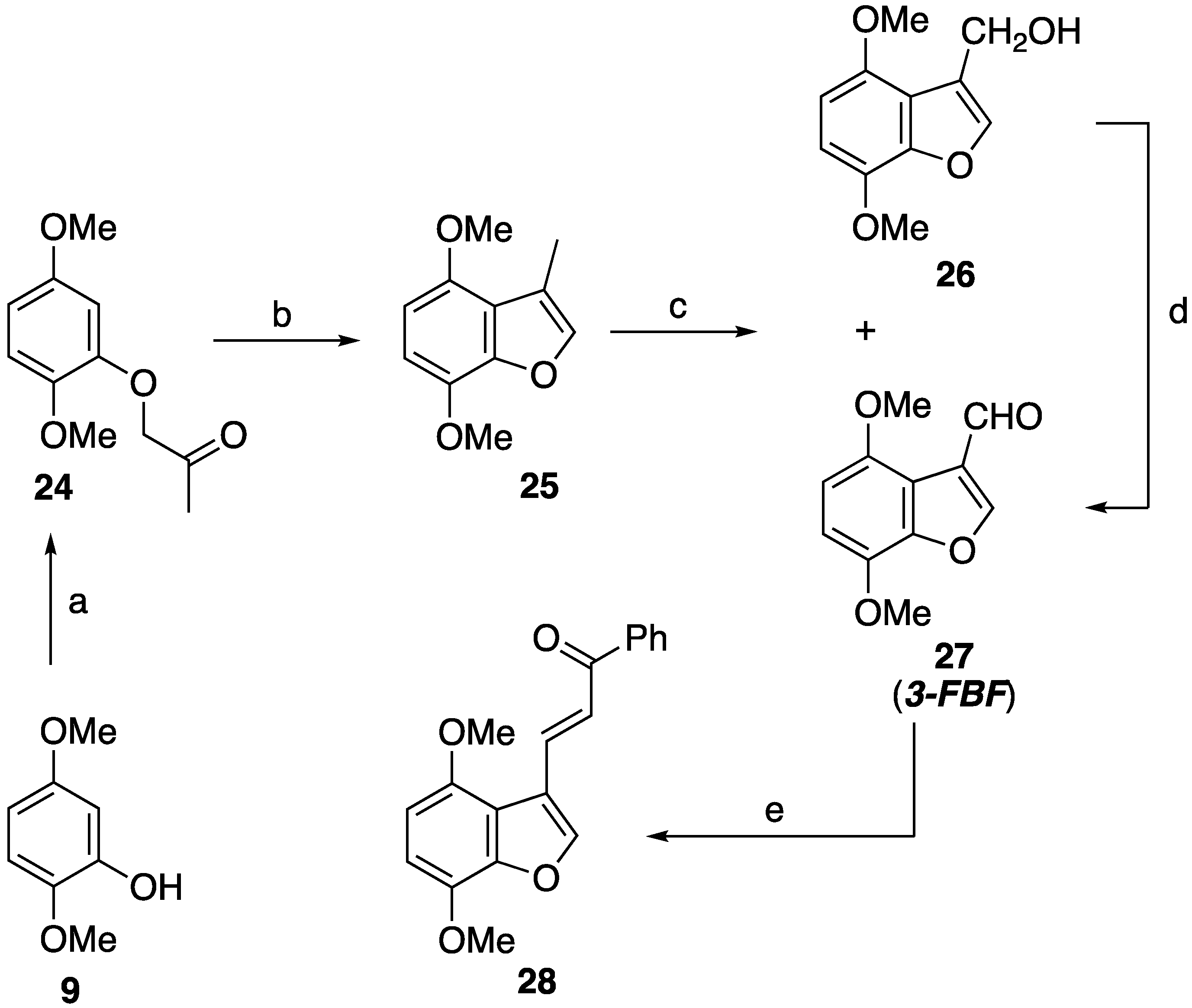
2.3.2. Synthetic Pathway to the Isomer 28
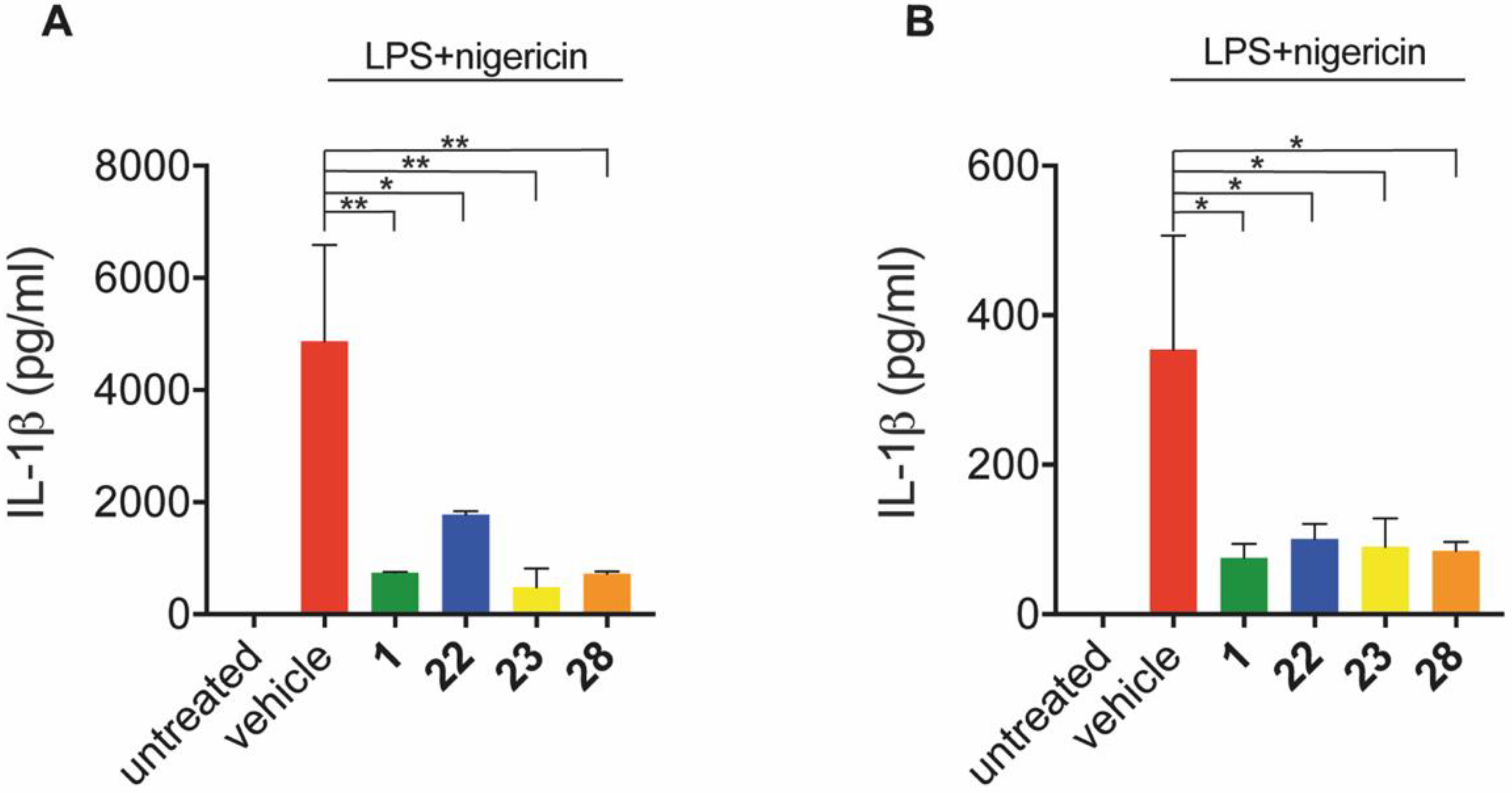
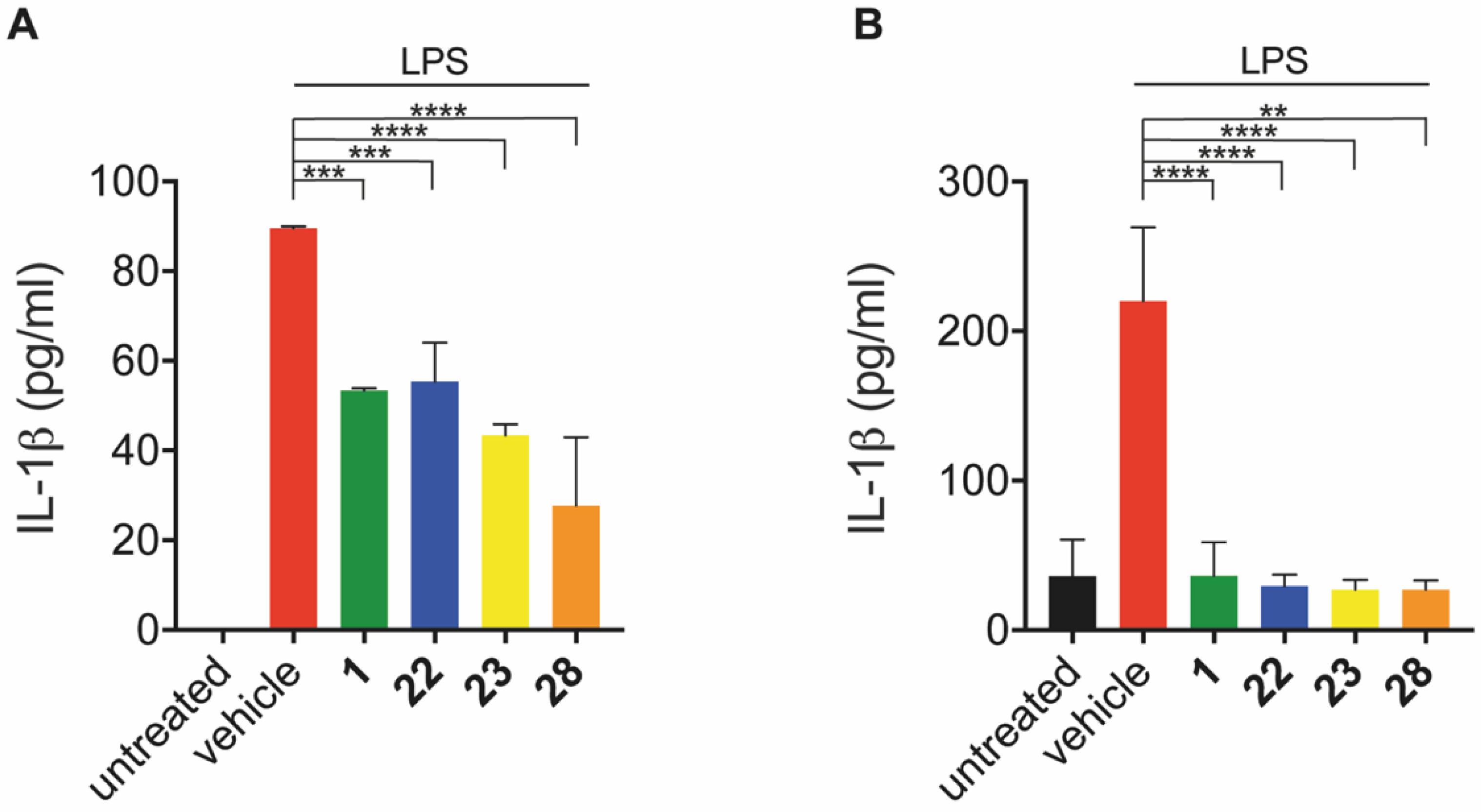
2.3.3. Inhibition of IL-1β Release In Vitro and In Vivo
3. Materials and Methods
3.1. Chemistry: Materials and General
3.1.1. Methyl 3-(4-Methoxy-4-oxobutanoyl)furan-2-carboxylate (3)
3.1.2. Methyl 3-(1,1,4-Trimethoxy-4-oxobutyl)furan-2-carboxylate (4)
3.1.3. Methyl 4-Hydroxy-7-methoxybenzofuran-5-carboxylate (5)
3.1.4. Methyl 4,7-Dimethoxybenzofuran-5-carboxylate (6)
3.1.5. 4,7-Dimethoxybenzofuran-5-carbaldehyde (7)
3.1.6. 2,5-Dimethoxyphenol (9)
3.1.7. 4-Bromo-2,5-dimethoxyphenol (10)
3.1.8. 1-Bromo-4-(2,2-dimethoxyethoxy)-2,5-dimethoxybenzene (11)
3.1.9. 5-Bromo-4,7-dimethoxybenzofuran (12) by Cyclization of (11)
3.1.10. 4,7-Dimethoxybenzofuran-5-carbaldehyde (7) via Halogen-Metal Exchange
3.1.11. 1-Phenylprop-2-en-1-one (13)
3.1.12. 2-(2,5-Dimethoxyphenoxy) tetrahydro-2H-pyran (14)
3.1.13. 2-Hydroxy-3,6-dimethoxybenzaldehyde (15)
3.1.14. 3-Bromo-6-hydroxy-2,5-dimethoxybenzaldehyde (16)
3.1.15. Ethyl 2-(4-bromo-2-formyl-3,6-dimethoxyphenoxy)acetate (17)
3.1.16. 5-Bromo-4,7-dimethoxybenzofuran (12) via Cyclization of (17)
3.1.17. Synthesis of (1) from (12) via Mizoroki-Heck
3.1.18. Synthesis of (1) from (7) via Wittig
3.1.19. (E)-3-(4,7-Dimethoxybenzofuran-2-yl)-1-phenylprop-2-en-1-one (22)
3.1.20. Synthesis of (22 and 1) from (20 and 7) via Wittig by Convectional Heating
3.1.21. (E)-3-(4,7-Dimethoxybenzofuran-6-yl)-1-phenylprop-2-en-1-one (23)
3.1.22. 2-(Dimethoxymethoxy)-1,4-dimethoxybenzene (18)
3.1.23. 4,7-Dimethoxybenzofuran (19)
3.1.24. 1-(2,5-Dimethoxyphenoxy)propan-2-one (24)
3.1.25. 4,7-Dimethoxy-3-methylbenzofuran (25)
3.1.26. (4,7-Dimethoxybenzofuran-3-yl)methanol (26), 4,7-Dimethoxybenzofuran-3-carbaldehyde (27)
3.1.27. 4,7-Dimethoxybenzofuran-3-carbaldehyde (27)
3.1.28. (E)-3-(4,7-Dimethoxybenzofuran-3-yl)-1-phenylprop-2-en-1-one (28)
3.1.29. Cell Cultures and Stimulation
3.1.30. In Vivo LPS Challenge
3.1.31. ELISA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Tehfe, M.-A.; Dumur, F.; Xiao, P.; Delgove, M.; Graff, B.; Fouassier, J.-P.; Gigmes, D.; Lalevée, J. Chalcone Derivatives as Highly Versatile Photoinitiators for Radical, Cationic, Thiol-ene and IPN Polymerization Reactions Upon Visible Lights. Polym. Chem. 2014, 5, 382–390. [Google Scholar] [CrossRef]
- Lee, S.-C.; Kang, N.-Y.; Park, S.-J.; Yun, S.-W.; Chandran, Y.; Chang, Y.-T. Development of a fluorescent chalcone library and its application in the discovery of a mouse embryonic stem cell probe. Chem. Commun. 2012, 48, 6681–6683. [Google Scholar] [CrossRef]
- Nowakowska, Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xing, C. Diverse Molecular Targets for Chalcones with Varied Bioactivities. Med. Chem. 2015, 5, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Funakoshi-Tago, M.; Tanabe, S.; Tago, K.; Itoh, H.; Mashino, T.; Sonoda, Y.; Kasahara, T. Licochalcone A Potently Inhibits Tumor Necrosis Factor α-Induced Nuclear Factor-kB Activation through the Direct Inhibition of IkB Kinase Complex Activation. Mol. Pharmacol. 2009, 76, 745–753. [Google Scholar] [CrossRef]
- Pandey, M.K.; Sandur, S.K.; Sung, B.; Sethi, G.; Kunnumakkara, A.B.; Aggarwal, B.B. Butein, a Tetrahydroxychalcone, Inhibits Nuclear Factor (NF)-kB and NF-kB-regulated Gene Expression through Direct Inhibition of IkBα Kinase β on Cysteine 179 Residue. J. Biol. Chem. 2007, 282, 17340–17350. [Google Scholar] [CrossRef]
- Wang, J.P.; Tsao, L.T.; Raung, S.L.; Lin, C.N. Investigation of the inhibitory effect of broussochalcone A on respiratory burst in neutrophils. Eur. J. Pharmacol. 1997, 320, 201–208. [Google Scholar] [CrossRef]
- Maccari, R.; Ottana, R. Targeting Aldose Reductase for the Treatment of Diabetes Complications and Inflammatory Diseases: New Insights and Future Directions. J. Med. Chem. 2015, 58, 2047–2067. [Google Scholar] [CrossRef]
- Aida, K.; Tawata, M.; Shindo, H.; Onaya, T.; Sasaki, H.; Yamaguchi, T.; Chin, M.; Mitsuhashi, H. Isoliquiritigenin: A New Aldose Reductase Inhibitor from Glycyrrhizae Radix. Planta Med. 1990, 56, 254–258. [Google Scholar] [CrossRef]
- Song, N.R.; Kim, J.E.; Park, J.S.; Kim, J.R.; Kang, H.; Lee, E.; Kang, Y.G.; Son, J.E.; Seo, S.G.; Heo, Y.S.; et al. Licochalcone A, a Polyphenol Present in Licorice, Suppresses UV-Induced COX-2 Expression by Targeting PI3K, MEK1, and B-Raf. Int. J. Mol. Sci. 2015, 16, 4453–4470. [Google Scholar] [CrossRef] [PubMed]
- Nyandoro, S.S.; Nkunya, M.H.H.; Josepha, C.C.; Odalo, J.O.; Sattler, I. New Glucopyranosylglyceryl-N-Octenyl Adipate and Bioactivity of Retro and Condensed Chalcones from Toussaintia orientalis. Tanz. J. Sci. 2012, 38, 108–126, eISSN: 2507–7961. Print ISSN 0856-1761. [Google Scholar]
- Lee, M.H.; Kim, J.Y.; Ryu, J.-H. Prenylflavones from Psoralea corylifolia Inhibit Nitric Oxide Synthase Expression through the Inhibition of I-kB-α Degradation in Activated Microglial Cells. Biol. Pharm. Bull. 2005, 28, 2253–2257. [Google Scholar] [CrossRef]
- Daikonya, A.; Katsuki, S.; Kitanaka, S. Antiallergic Agents from Natural Sources 9. Inhibition of Nitric Oxide Production by Novel Chalcone Derivatives from Mallotus philippinensis (Euphorbiaceae). Chem. Pharm. Bull. 2004, 52, 1326–1329. [Google Scholar] [CrossRef]
- Chen, H.; Chen, X.; Sun, P.; Wu, D.; Yue, H.; Pan, J.; Li, X.; Zhang, C.; Wu, X.; Hua, L.; et al. Discovery of dronedarone and its analogues as NLRP3 inflammasome inhibitors with potent anti-inflammation activity. Bioorg. Med. Chem. Lett. 2021, 46, 128160. [Google Scholar] [CrossRef]
- Zhang, C.; Yue, H.; Sun, P.; Hua, L.; Liang, S.; Ou, Y.; Wu, D.; Wu, X.; Chen, H.; Hao, Y.; et al. Discovery of chalcone analogues as novel NLRP3 inflammasome inhibitors with potent anti-inflammation activities. Eur. J. Med. Chem. 2021, 219, 113417. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.R.; Lim, H.; Lee, J.H.; Park, H.; Kim, H.P. Interruption of Helicobacter pylori-Induced NLRP3 Inflammasome Activation by Chalcone Derivatives. Biomol. Ther. 2021, 29, 410–418. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, G.; Gao, Y.; Zhan, X.; Qin, N.; Fu, S.; Li, R.; Niu, M.; Wang, J.; Liu, Y.; et al. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm. Sin. B 2019, 9, 734–744. [Google Scholar] [CrossRef]
- Aybe, S.I.; Furuya, T. Studies on Plant Tissue Cultures. Part 36. Biosynthesis of a Retrochalcone, Echinatin, and Other Flavonoids in the Cultured Cells of Glycyrrhiza echinate. A New Route to a Chalcone with Transposed A- and B-Rings. J. Chem. Soc. Perkin Trans. 1982, 1, 2725–2734. [Google Scholar] [CrossRef]
- Aybe, S.I.; Furuya, T. Biosynthesis of a Retrochalcone, Echinatin: A Feeding Study with Advanced Precursors. Tetrahedron Lett. 1981, 22, 2097–2098. [Google Scholar] [CrossRef]
- Ayabe, S.I.; Yoshikawa, T.; Kobayashi, M.; Furuya, T. Biosynthesis of a Retrochalcone, Echinatin: Involvement of O-Methyl Transferase to Licodione. Phytochemistry 1980, 19, 2331–2336. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, M.; Tang, M.-H.; Xue, L.-L.; Zhang, R.-J.; Liu, L.; Ni, H.-F.; Cai, X.-Y.; Kuang, S.; Hong, F.; et al. Flavonoids with Inhibitory Effects on NLRP3 Inflammasome Activation from Millettia velutina. J. Nat. Prod. 2020, 83, 2950–2959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hong, F.; Zhao, M.; Cai, X.; Jiang, X.; Ye, N.; Su, K.; Li, N.; Tang, M.; Ma, X.; et al. New Highly Potent NLRP3 Inhibitors: Furanochalcone Velutone F Analogues. ACS Med. Chem. Lett. 2022, 13, 560–569. [Google Scholar] [CrossRef]
- Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef]
- Miao, Y.H.; Hu, Y.H.; Yang, J.; Liu, T.; Sun, J.; Wang, X.J. Natural source, bioactivity and synthesis of benzofuran derivatives. RSC Adv. 2019, 9, 27510–27540. [Google Scholar] [CrossRef]
- Chand, K.; Hiremathad, A.; Singh, M.; Santos, M.A.; Keri, R.S. A review on antioxidant potential of bioactive heterocycle benzofuran: Natural and synthetic derivatives. Pharm. Rep. 2017, 69, 281–295. [Google Scholar] [CrossRef]
- Radadiya, A.; Shah, A. Bioactive benzofuran derivatives: An insight on lead developments, radioligands and advances of the last decade. Eur. J. Med. Chem. 2015, 97, 356–376. [Google Scholar] [CrossRef]
- Khanam, H. Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef]
- Schwaid, A.G.; Spencer, K.B. Strategies for Targeting the NLRP3 Inflammasome in the Clinical and Preclinical Space. J. Med. Chem. 2021, 64, 101–122. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, A.; Lv, J.; Zhang, Q.; Ran, Y.; Wei, C.; Wu, J. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur. J. Med. Chem. 2020, 185, 1118222. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; de Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef]
- Goyal, K.; Kaur, R.; Goyal, A.; Awasthi, R. Chalcones: A review on synthesis and pharmacological activities. J. Appl. Pharm. Sci. 2021, 11 (Suppl. S1), 1–14. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, F.; Huang, S.; Zhao, Z.; Mao, H.; Qin, W. Organocatalytic Enantioselective Selenosulfonylation of a C−C Double Bond to Form Two Stereogenic Centers in an Aqueous Medium. J. Org. Chem. 2019, 84, 8100–8111. [Google Scholar] [CrossRef]
- Gammill, R.B. Synthetic Routes to Benzofurans and Benzothiophenes and Intermediates Therefor. U.S. Patent 4,609,739, 2 September 1986. [Google Scholar]
- Gammill, R.B. Preparing 4,7-Dialkoxybenzofurans, and Intermediates Used Therein. European Patent 95,835, 7 December 1983. [Google Scholar]
- Guzman, J.A.; Mendoza, V.; Garcia, E.; Garibay, C.F.; Olivares, L.Z.; Maldonado, L.A. Baeyer-Villiger Oxidation of β-Aryl Substituted Unsaturated Carbonyl Compounds with Hydrogen Peroxide and Catalytic Selenium Dioxide. Synth. Commun. 1995, 25, 2121–2133. [Google Scholar] [CrossRef]
- Pang, Y.; An, B.; Lou, L.; Zhang, J.; Yan, J.; Huang, L.; Li, X.; Yin, S. Design, Synthesis, and Biological Evaluation of Novel Selenium- Containing Isocombretastatins and Phenstatins as Antitumor Agents. J. Med. Chem. 2017, 60, 7300–7314. [Google Scholar] [CrossRef]
- Ma, W.; Huang, J.; Huang, X.; Meng, S.; Yang, Z.; Li, C.; Wang, Y.; Qi, T.; Li, B. Direct construction of 2,3-unsubstituted benzofurans and benzothiophenes via a metal-free catalyzed intramolecular Friedel-Crafts reaction. Org. Chem. Front. 2019, 6, 493–497. [Google Scholar] [CrossRef]
- Liu, H.; Ge, L.; Wang, D.; Chen, N.; Feng, C. Photoredox-Coupled F-Nucleophilic Addition: Allylation of gem-Difluoroalkenes, Angew. Chem. Int. Ed. 2019, 58, 3918–3922. [Google Scholar] [CrossRef]
- Song, S.-Y.; Lu, H.-L.; Wang, G.-F.; Yang, Y.-Q.; Huang, Y.-S. An improved and scale-up synthesis of 6-hydroxybenzofuran. Res. Chem. Intermed. 2016, 42, 4433–4442. [Google Scholar] [CrossRef]
- Bunce, R.A.; Moore, J.D. Tetrahydropyranyloxy-Directed ortho Lithiation of Aromatic Systems. Synthesis of o-Hydroxycinnamate Esters from Phenols. Org. Prep. Proc. Int. 1997, 29, 293–299. [Google Scholar] [CrossRef]
- Wriede, U.; Fernandez, M.; West, K.F.; Harcour, D.; Moore, H.W. Synthesis of Halodimethoxy-1,2-benzoquinones. J. Org. Chem. 1987, 52, 4485–4489. [Google Scholar] [CrossRef]
- Ma, T.; Fu, X.; Kee, C.W.; Zong, L.; Pan, Y.; Huang, K.-W.; Tan, C.-H. Pentanidium-Catalyzed Enantioselective Phase-Transfer Conjugate Addition Reactions. J. Am. Chem. Soc. 2011, 133, 2828–2831. [Google Scholar] [CrossRef]
- Mikami, S.; Kitamura, S.; Negoro, N.; Sasaki, S.; Suzuki, M.; Tsujihata, Y.; Miyazaki, T.; Ito, R.; Suzuki, N.; Miyazaki, J.; et al. Discovery of Phenylpropanoic Acid Derivatives Containing Polar Functionalities as Potent and Orally Bioavailable G Protein-Coupled Receptor 40 Agonists for the Treatment of Type 2 Diabetes. J. Med. Chem. 2012, 53, 3756–3776. [Google Scholar] [CrossRef]
- Sayson, L.V.; Custodio, R.J.P.; Ortiz, D.M.; Lee, H.J.; Kim, M.; Jeong, Y.; Lee, Y.S.; Kim, H.J.; Cheong, J.H. The potential rewarding and reinforcing effects of the substituted benzofurans 2-EAPB and 5-EAPB in rodents. Eur. J. Pharmacol. 2020, 885, 173527. [Google Scholar] [CrossRef]
- Sun, N.; Huang, P.; Wang, Y.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. Zeolite-catalyzed synthesis of 2,3-unsubstituted benzo[b]furans via the intramolecular cyclization of 2-aryloxyacetaldehyde acetals. Tetrahedron 2015, 71, 4835–4841. [Google Scholar] [CrossRef]
- Zaidlewicz, M.; Chechlowska, A.; Prewysz-Kwinto, A. Enantioselective Synthesis of 2- and 3-benzofuryl β-amino alcohols. Heterocycles 2001, 55, 569–577. [Google Scholar] [CrossRef]
- Ando, K.; Kawamura, Y.; Akai, Y.; Kunitomo, J.I.; Yokomizo, T.; Yamashita, M.; Ohta, S.; Ohishi, T.; Ohishi, Y. Preparation of 2-, 3-, 4- and 7-(2-alkylcarbamoyl-1-alkylvinyl)benzo[b]furans and their BLT1 and/or BLT2 inhibitory activities. Org. Biomol. Chem. 2008, 6, 296–307. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Boncompagni, C.; Borghi, C.; Campagnaro, A.; Marchetti, F.; Anania, G.; Greco, P.; Fiorica, F.; Pinton, P.; et al. Targeting the NLRP3 inflamasome as a new therapeutic option for overcoming cancer. Cancers 2021, 13, 2297. [Google Scholar] [CrossRef]
- Ying, W.; Cheruku, P.S.; Bazer, F.W.; Safe, S.H.; Zhou, B. Investigation of macrophage polarization using bone marrow derived macrophages. J. Vis. Exp. 2013, 76, 50323. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Ventura, T.; Perrone, M.; Missiroli, S.; Pinton, P.; Marchetti, P.; Strazzabosco, G.; Turrin, G.; Illuminati, D.; Cristofori, V.; Fantinati, A.; et al. Synthesis and NLRP3-Inflammasome Inhibitory Activity of the Naturally Occurring Velutone F and of Its Non-Natural Regioisomeric Chalconoids. Int. J. Mol. Sci. 2022, 23, 8957. https://doi.org/10.3390/ijms23168957
De Ventura T, Perrone M, Missiroli S, Pinton P, Marchetti P, Strazzabosco G, Turrin G, Illuminati D, Cristofori V, Fantinati A, et al. Synthesis and NLRP3-Inflammasome Inhibitory Activity of the Naturally Occurring Velutone F and of Its Non-Natural Regioisomeric Chalconoids. International Journal of Molecular Sciences. 2022; 23(16):8957. https://doi.org/10.3390/ijms23168957
Chicago/Turabian StyleDe Ventura, Tiziano, Mariasole Perrone, Sonia Missiroli, Paolo Pinton, Paolo Marchetti, Giovanni Strazzabosco, Giulia Turrin, Davide Illuminati, Virginia Cristofori, Anna Fantinati, and et al. 2022. "Synthesis and NLRP3-Inflammasome Inhibitory Activity of the Naturally Occurring Velutone F and of Its Non-Natural Regioisomeric Chalconoids" International Journal of Molecular Sciences 23, no. 16: 8957. https://doi.org/10.3390/ijms23168957