
Glioblastoma Extracellular Vesicle-Specific Peptides Inhibit EV-Induced Neuronal Cytotoxicity

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Characterization of Brain Tumor Plasma-Derived Extracellular Vesicles
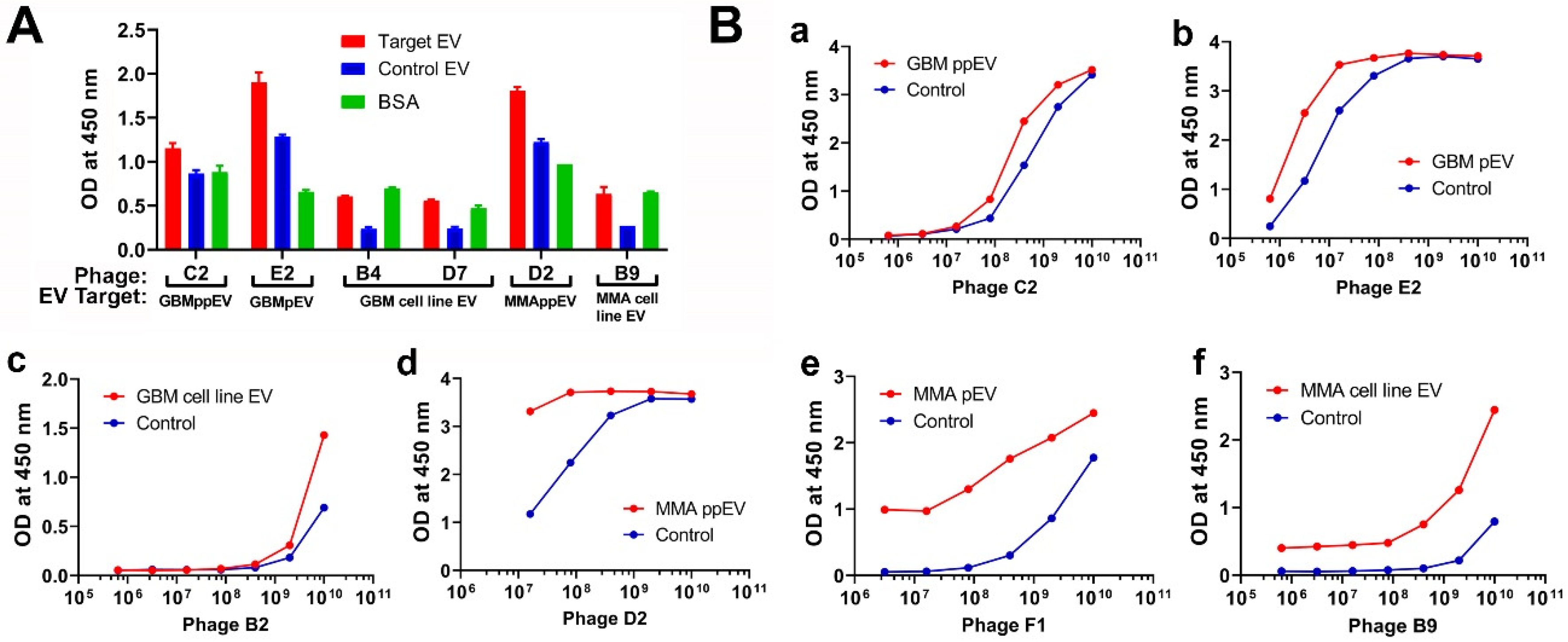
2.2. Screening Phage-Displayed Random Peptide Libraries with Brain Tumor EVs Identified High-Affinity Phage Peptides with Identical and Homologous Sequences
2.3. Phage ELISA Assays Demonstrated the Specific Bindings of Phage Peptides to Corresponding Panning Brain Tumor EVs
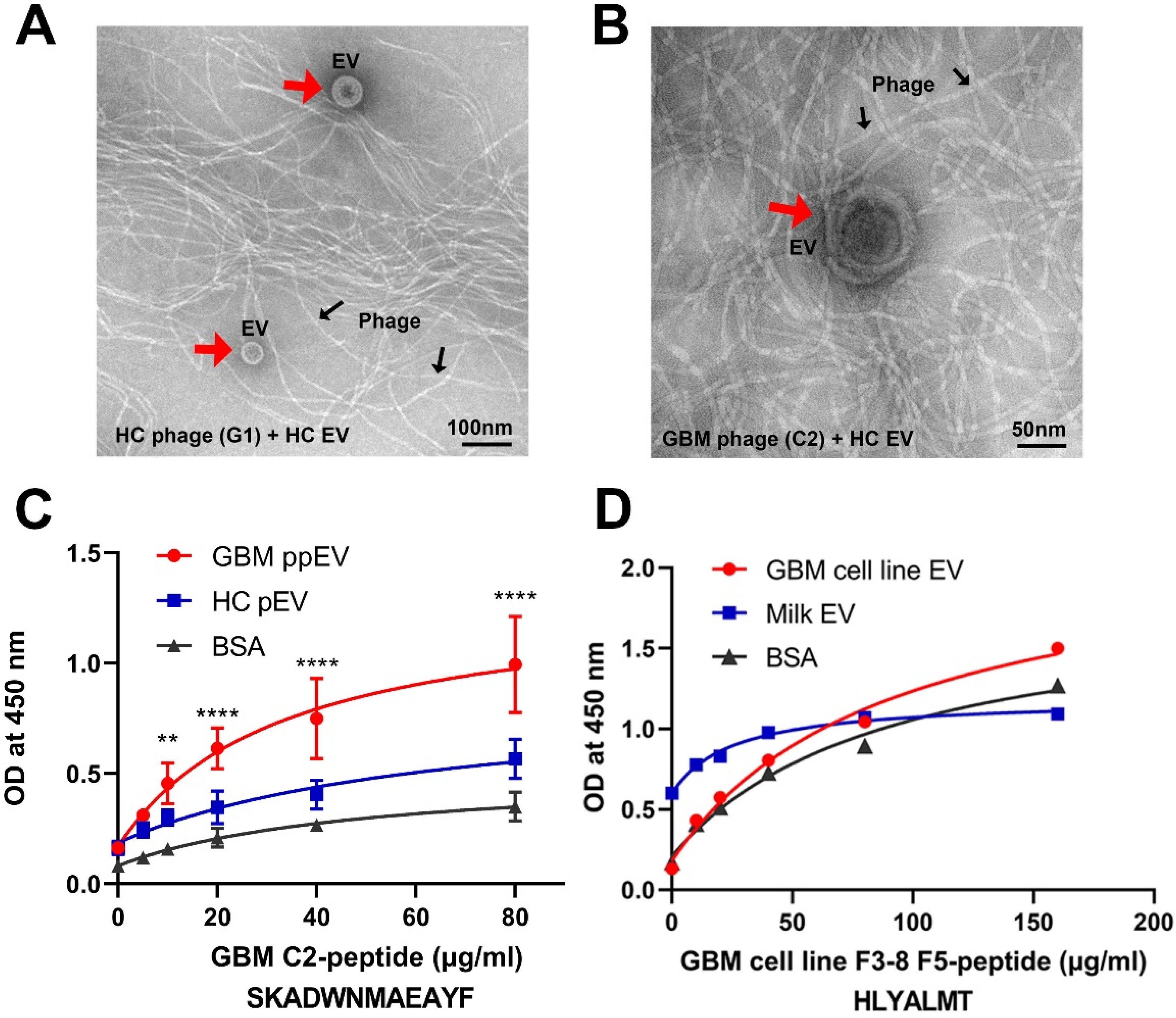
2.4. Transmission Electron Microscopy Analysis of PHAGE–EV Co-Precipitation
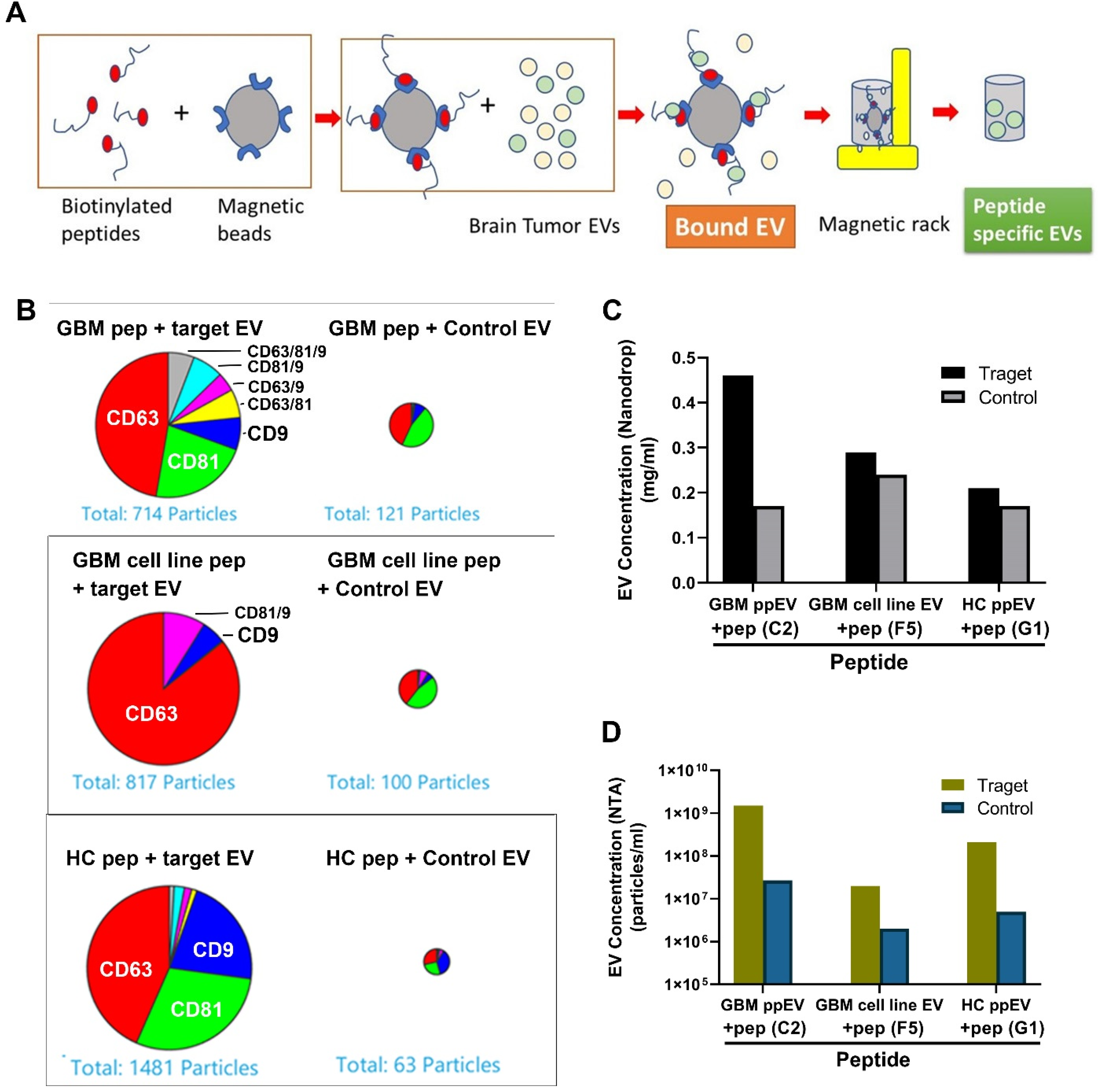
2.5. Affinity Binding Analysis of Synthetic Peptides Confirmed Specific Bindings to Brain Tumor EVs
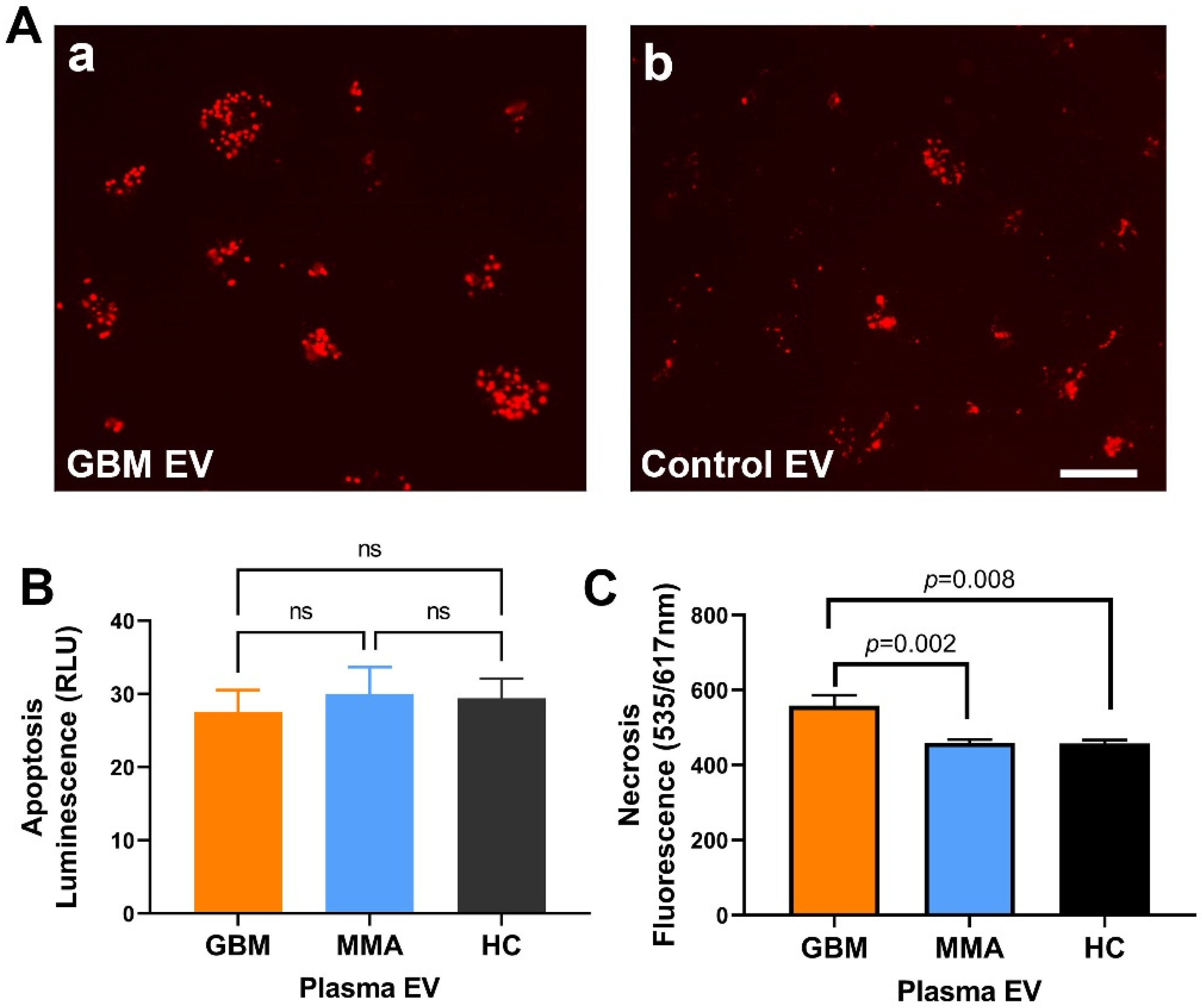
2.6. GBM EV Caused Necrotic Cell Death in Neurons
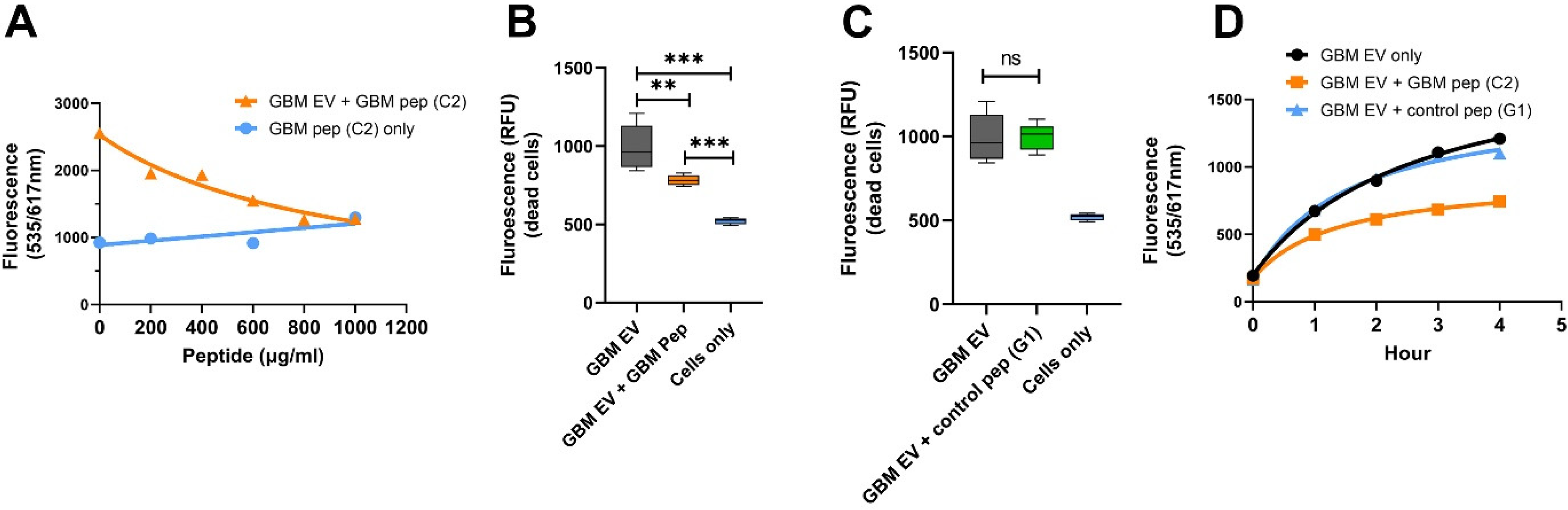
2.7. GBM EV-Specific Peptides Inhibit Neuronal Cytotoxicity Caused by the GBM EVs
3. Discussion
4. Materials and Methods
4.1. Human Samples
4.2. Cell Lines
4.3. Extracellular Vesicle Isolation Using the Ultracentrifugation Method
4.4. Extracellular Vesicle Isolation Using ExoEasy and ExoQuick Commercial Kits
4.5. EV Characterization
4.6. Western Blots
4.7. Phage-Displayed Random Peptide Library Screening
4.7.1. Phage Titering, Amplification, and DNA Sequencing
4.7.2. DNA Sequencing of Individual Phages
4.8. Phage ELISA with Anti-M13 Antibody
4.9. Peptide Synthesis
4.10. Synthetic Peptide ELISA
4.11. Negative Staining of EVs and Visualization via Transmission Electron Microscopy (TEM)
4.12. Phage–EV Co-Precipitation
4.13. Peptide Affinity Precipitation of GBM EVs
4.14. Assessing EV Cytotoxicity in Neuroblastoma SH-SY5Y Cells
4.15. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (≥2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Huntoon, K.; Toland, A.M.S.; Dahiya, S. Meningioma: A Review of Clinicopathological and Molecular Aspects. Front. Oncol. 2020, 10, 579599. [Google Scholar] [CrossRef]
- Preusser, M.; Brastianos, P.; Mawrin, C. Advances in meningioma genetics: Novel therapeutic opportunities. Nat. Rev. Neurol. 2018, 14, 106–115. [Google Scholar] [CrossRef]
- Budnik, V.; Ruiz-Cañada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzás, E.I.; Tóth, E.; Sódar, B.W.; Szabó-Taylor, K. Molecular interactions at the surface of extracellular vesicles. Semin. Immunopathol. 2018, 40, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Cappariello, A.; Rucci, N. Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone. Int. J. Mol. Sci. 2019, 20, 4805. [Google Scholar] [CrossRef] [Green Version]
- Logozzi, M.; Spugnini, E.; Mizzoni, D.; Di Raimo, R.; Fais, S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019, 38, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hasan, H.; Sohal, I.S.; Soto-Vargas, Z.; Byappanahalli, A.M.; Humphrey, S.E.; Kubo, H.; Kitdumrongthum, S.; Copeland, S.; Tian, F.; Chairoungdua, A.; et al. Extracellular vesicles released by non-small cell lung cancer cells drive invasion and permeability in non-tumorigenic lung epithelial cells. Sci. Rep. 2022, 12, 972. [Google Scholar] [CrossRef] [PubMed]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos. Trans. R. Soc. B Biol. Sci. 2017, 373, 20160477. [Google Scholar] [CrossRef] [Green Version]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2018, 56, 4566–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.L.N.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W. HSP90 and Immune Modulation in Cancer. Adv. Cancer Res. 2016, 129, 191–224. [Google Scholar] [CrossRef]
- Graner, M.W.; Schnell, S.; Olin, M.R. Tumor-derived exosomes, microRNAs, and cancer immune suppression. Semin. Immunopathol. 2018, 40, 505–515. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Veerman, R.E.; Teeuwen, L.; Czarnewski, P.; Akpinar, G.G.; Sandberg, A.; Cao, X.; Pernemalm, M.; Orre, L.M.; Gabrielsson, S.; Eldh, M. Molecular evaluation of five different isolation methods for extracellular vesicles reveals different clinical applicability and subcellular origin. J. Extracell. Vesicles 2021, 10, e12128. [Google Scholar] [CrossRef]
- Leoni, G.; Neumann, P.-A.; Kamaly, N.; Quiros, M.; Nishio, H.; Jones, H.R.; Sumagin, R.; Hilgarth, R.S.; Alam, A.; Fredman, G.; et al. Annexin A1–containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J. Clin. Investig. 2015, 125, 1215–1227. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Tateno, H.; Kuno, A.; Yabe, R.; Hirabayashi, J. Directed Evolution of Lectins with Sugar-binding Specificity for 6-Sulfo-galactose. J. Biol. Chem. 2012, 287, 20313–20320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowlands, D.; Sugahara, K.; Kwok, J.C.F. Glycosaminoglycans and Glycomimetics in the Central Nervous System. Molecules 2015, 20, 3527–3548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.M.; Matsumoto, M.; Omi, H.; Kobayashi, T.; Nakamura, A.; Kishi, H.; Kobayashi, S.; Takagi, T. Interaction of peptide-bound beads with lipopolysaccharide and lipoproteins. J. Microbiol. Methods 2014, 100, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Matsumoto, M.; Yamamoto, A.; Ochiai, M.; Horiuchi, Y.; Niwa, M.; Omi, H.; Kobayashi, T.; Takagi, T. Molecular design of LPS-binding peptides. J. Microbiol. Methods 2010, 83, 153–155. [Google Scholar] [CrossRef]
- Liu, J.K.; Teng, Q.; Garrity-Moses, M.; Federici, T.; Tanase, D.; Imperiale, M.J.; Boulis, N.M. A novel peptide defined through phage display for therapeutic protein and vector neuronal targeting. Neurobiol. Dis. 2005, 19, 407–418. [Google Scholar] [CrossRef]
- Miura, Y.; Sasao, Y.; Kamihira, M.; Sakaki, A.; Iijima, S.; Kobayashi, K. Peptides binding to a Gb3 mimic selected from a phage library. Biochim. Biophys. Acta Gen. Subj. 2004, 1673, 131–138. [Google Scholar] [CrossRef]
- Thomas, C.J.; Sharma, S.; Kumar, G.; Visweswariah, S.S.; Surolia, A. Biopanning of endotoxin-specific phage displayed peptides. Biochem. Biophys. Res. Commun. 2003, 307, 133–138. [Google Scholar] [CrossRef]
- Vetter, S.W. Phage Display Selection of Peptides that Target Calcium-Binding Proteins. Methods Mol. Biol. 2012, 963, 215–235. [Google Scholar] [CrossRef]
- Wu, C.-H.; Liu, I.-J.; Lu, R.-M.; Wu, H.-C. Advancement and applications of peptide phage display technology in biomedical science. J. Biomed. Sci. 2016, 23, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Gilden, D.H.; Ritchie, A.M.; Burgoon, M.P.; Keays, K.M.; Owens, G.P. Specificity of recombinant antibodies generated from multiple sclerosis cerebrospinal fluid probed with a random peptide library. J. Neuroimmunol. 2006, 172, 121–131. [Google Scholar] [CrossRef]
- Yu, X.; Gilden, D.; Schambers, L.; Barmina, O.; Burgoon, M.; Bennett, J.; Owens, G. Peptide reactivity between multiple sclerosis (MS) CSF IgG and recombinant antibodies generated from clonally expanded plasma cells in MS CSF. J. Neuroimmunol. 2011, 233, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graner, M.; Pointon, T.; Manton, S.; Green, M.; Dennison, K.; Davis, M.; Braiotta, G.; Craft, J.; Edwards, T.; Polonsky, B.; et al. Oligoclonal IgG antibodies in multiple sclerosis target patient-specific peptides. PLoS ONE 2020, 15, e0228883. [Google Scholar] [CrossRef]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishibori, M.; Cham, B.; McNicol, A.; Shalev, A.; Jain, N.; Gerrard, J.M. The protein CD63 is in platelet dense granules, is deficient in a patient with Hermansky-Pudlak syndrome, and appears identical to granulophysin. J. Clin. Investig. 1993, 91, 1775–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azorsa, D.O.; Hyman, J.A.; Hildreth, J.E. CD63/Pltgp40: A platelet activation antigen identical to the stage-specific, melanoma-associated antigen ME491. Blood 1991, 78, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Barmina, O.; Burgoon, M.; Gilden, D. Identification of measles virus epitopes using an ultra-fast method of panning phage-displayed random peptide libraries. J. Virol. Methods 2009, 156, 169–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarovni, N.; Corrado, A.; Guazzi, P.; Zocco, D.; Lari, E.; Radano, G.; Muhhina, J.; Fondelli, C.; Gavrilova, J.; Chiesi, A. Integrated isolation and quantitative analysis of exosome shuttled proteins and nucleic acids using immunocapture approaches. Methods 2015, 87, 46–58. [Google Scholar] [CrossRef]
- Klingeborn, M.; Skiba, N.P.; Stamer, W.D.; Rickman, C.B. Isolation of Retinal Exosome Biomarkers from Blood by Targeted Immunocapture. Retin. Degener. Dis. 2019, 1185, 21–25. [Google Scholar] [CrossRef]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef]
- Greening, D.W.; Xu, R.; Ji, H.; Tauro, B.J.; Simpson, R.J. A Protocol for Exosome Isolation and Characterization: Evaluation of Ultracentrifugation, Density-Gradient Separation, and Immunoaffinity Capture Methods. Methods Mol. Biol. 2015, 1295, 179–209. [Google Scholar] [CrossRef]
- Yamauchi, M.; Shimizu, K.; Rahman, M.; Ishikawa, H.; Takase, H.; Ugawa, S.; Okada, A.; Inoshima, Y. Efficient method for isolation of exosomes from raw bovine milk. Drug Dev. Ind. Pharm. 2018, 45, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Boggiano, C.; Reixach, N.; Pinilla, C.; Blondelle, S.E. Successful identification of novel agents to control infectious diseases from screening mixture-based peptide combinatorial libraries in complex cell-based bioassays. Biopolymers 2003, 71, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Reilley, K.J.; Giulianotti, M.; Dooley, C.T.; Nefzi, A.; McLaughlin, J.P.; Houghten, R.A. Identification of Two Novel, Potent, Low-Liability Antinociceptive Compounds from the Direct In Vivo Screening of a Large Mixture-Based Combinatorial Library. AAPS J. 2010, 12, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denholt, C.L.; Hansen, P.R.; Pedersen, N.; Poulsen, H.S.; Gillings, N.; Kjaer, A. Identification of novel peptide ligands for the cancer-specific receptor mutation EFGRvIII using a mixture-based synthetic combinatorial library. Biopolymers 2008, 91, 201–206. [Google Scholar] [CrossRef]
- Pinilla, C.; Giulianotti, M.A.; Santos, R.G.; Houghten, R.A. Identification of B Cell and T Cell Epitopes Using Synthetic Peptide Combinatorial Libraries. Curr. Protoc. Immunol. 2022, 2, e378. [Google Scholar] [CrossRef] [PubMed]
- Sandomenico, A.; Caporale, A.; Doti, N.; Cross, S.; Cruciani, G.; Chambery, A.; De Falco, S.; Ruvo, M. Synthetic Peptide Libraries: From Random Mixtures to In Vivo Testing. Curr. Med. Chem. 2020, 27, 997–1016. [Google Scholar] [CrossRef]
- Hellwinkel, J.E.; Redzic, J.S.; Harland, T.A.; Gunaydin, D.; Anchordoquy, T.J.; Graner, M.W. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro-Oncology 2015, 18, 497–506. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, W.; Craft, J.; Ojemann, A.; Bergen, L.; Graner, A.; Gonzales, A.; He, Q.; Kopper, T.; Smith, M.; Graner, M.W.; et al. Glioblastoma Extracellular Vesicle-Specific Peptides Inhibit EV-Induced Neuronal Cytotoxicity. Int. J. Mol. Sci. 2022, 23, 7200. https://doi.org/10.3390/ijms23137200
Zhou W, Craft J, Ojemann A, Bergen L, Graner A, Gonzales A, He Q, Kopper T, Smith M, Graner MW, et al. Glioblastoma Extracellular Vesicle-Specific Peptides Inhibit EV-Induced Neuronal Cytotoxicity. International Journal of Molecular Sciences. 2022; 23(13):7200. https://doi.org/10.3390/ijms23137200
Chicago/Turabian StyleZhou, Wenbo, Julia Craft, Alex Ojemann, Luke Bergen, Arin Graner, Aitana Gonzales, Qianbin He, Timothy Kopper, Marie Smith, Michael W. Graner, and et al. 2022. "Glioblastoma Extracellular Vesicle-Specific Peptides Inhibit EV-Induced Neuronal Cytotoxicity" International Journal of Molecular Sciences 23, no. 13: 7200. https://doi.org/10.3390/ijms23137200