
Melatonin and the Brain–Heart Crosstalk in Neurocritically Ill Patients—From Molecular Action to Clinical Practice

 , , , and
, , , and 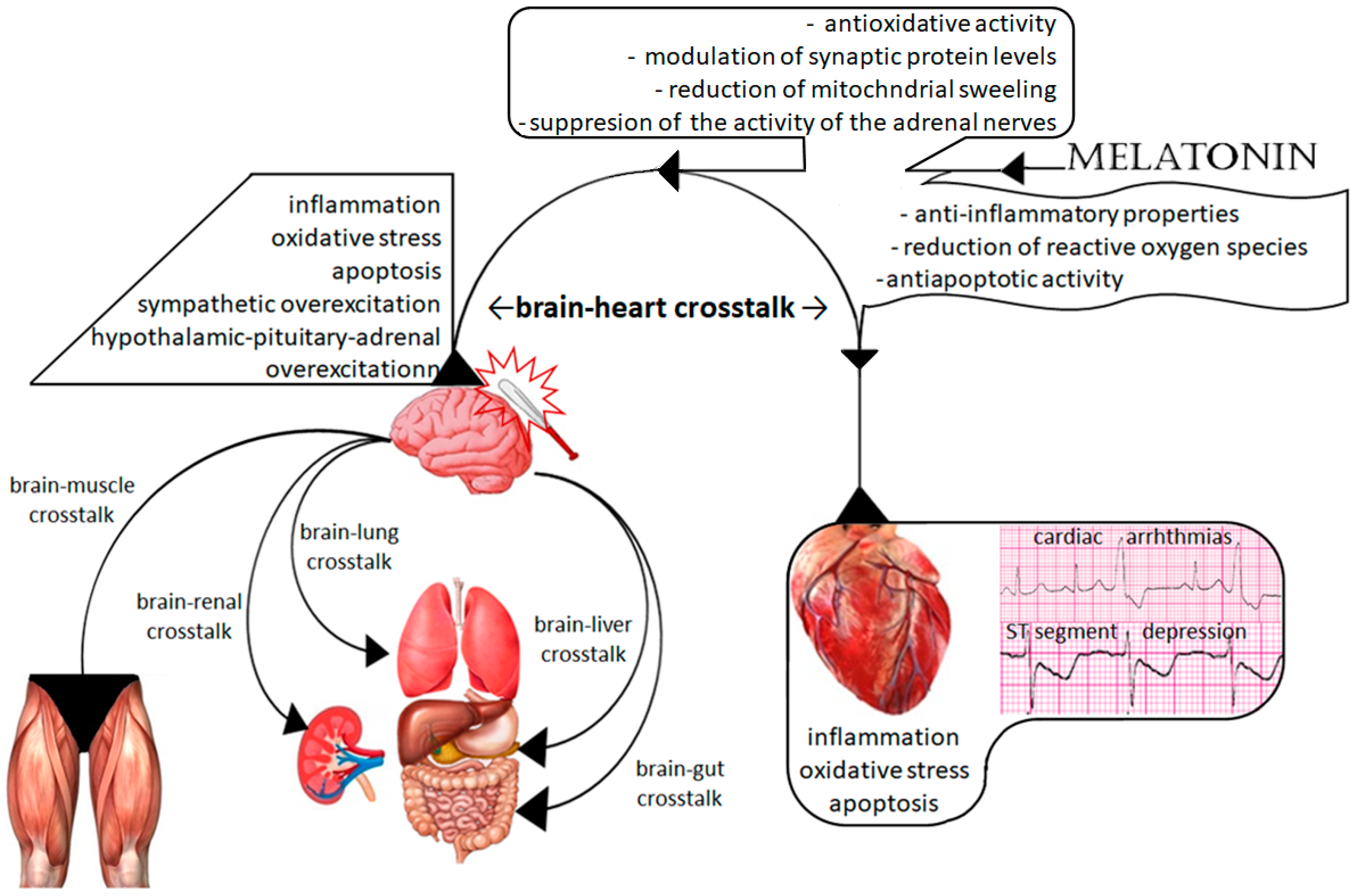{kind=link}
{kind=link}
Abstract
:1. Introduction
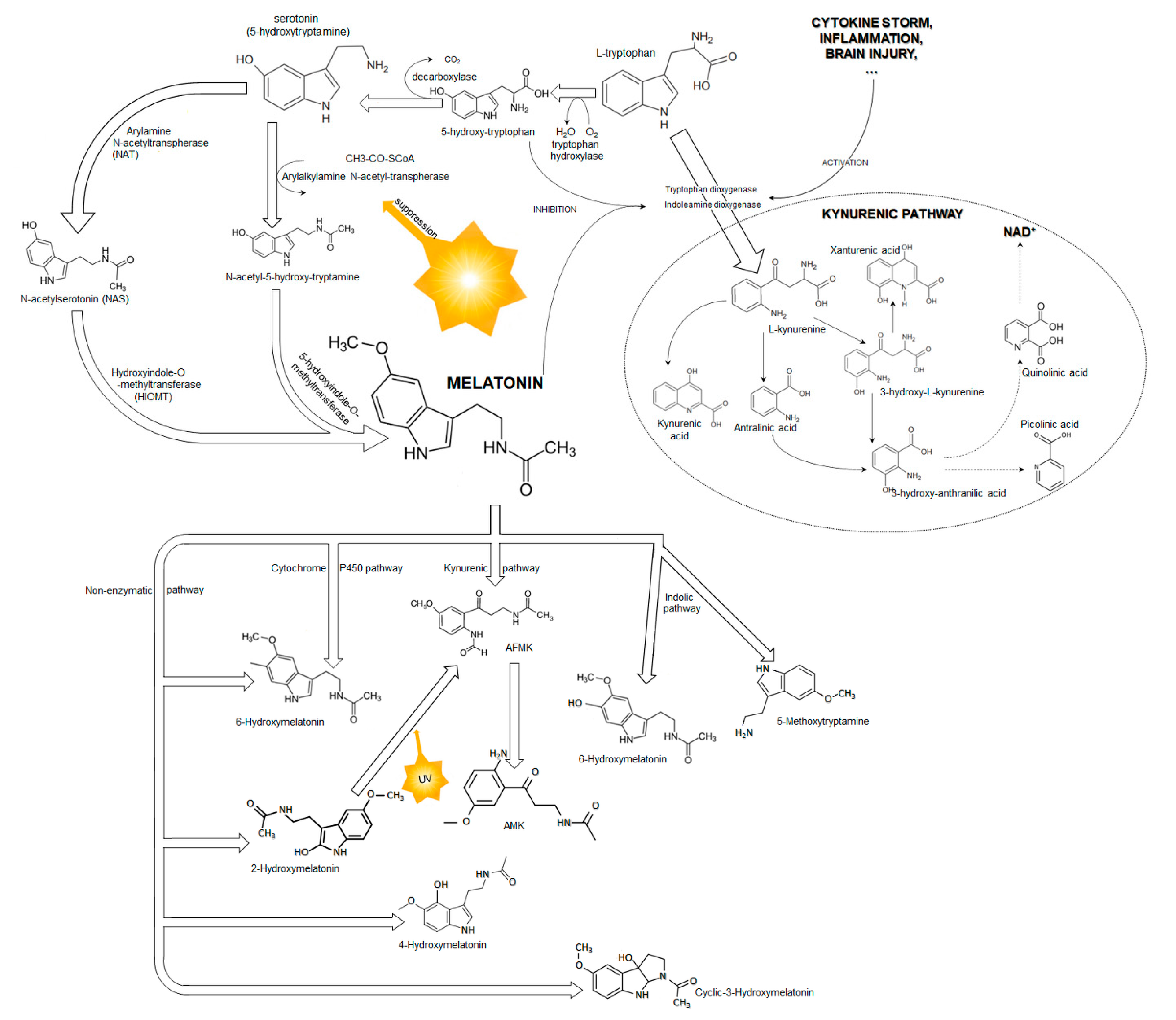
1.1. Melatonin
1.2. Brain Injury and Melatonin
1.3. The Brain–Heart Interaction
2. Possible Molecular Mechanisms of Melatonin Protection
2.1. Cerebral Effect of Melatonin on TBI-Related Cardiac Dysfunction
2.2. Direct Cardiac Protection
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maas, A.I.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picetti, E.; Rossi, S.; Abu-Zidan, F.M.; Ansaloni, L.; Armonda, R.; Baiocchi, G.L.; Bala, M.; Balogh, Z.J.; Berardino, M.; Biffl, W.L.; et al. WSES consensus conference guidelines: Monitoring and management of severe adult traumatic brain injury patients with polytrauma in the first 24 hours. World J. Emerg. Surg. 2019, 29, 53. [Google Scholar] [CrossRef] [PubMed]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the management of severe traumatic brain injury fourth edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Osier, N.; McGreevy, E.; Pham, L.; Puccio, A.; Ren, D.; Conley, Y.P.; Alexander, S.; Dixon, C.E. Melatonin as a therapy for traumatic brain injury: A review of published evidence. Int. J. Mol. Sci. 2018, 19, 1539. [Google Scholar] [CrossRef] [Green Version]
- Yates, N.; Gunn, A.J.; Bennet, L.; Dhillon, S.K.; Davidson, J.O. Preventing brain injury in the preterm infant—Current controversies and potential therapies. Int. J. Mol. Sci. 2021, 22, 1671. [Google Scholar] [CrossRef]
- Blum, B.; Kaushal, S.; Khan, S.; Kim, J.H.; Alvarez Villaba, C.L. Melatonin in traumatic brain injury and cognition. Cureus 2021, 13, 17776. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Ok Kim, M. Neurological enhancement effects of melatonin against brain injury-induced oxidative stress, neuroinflammation and neurodegeneration via AMPK/CREB signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef] [Green Version]
- Baltatu, O.C.; Senar, S.; Campos, L.A.; Cipolla-Neto, J. Cardioprotective melatonin: Translating from proof-of-concept studies to therapeutic use. Int. J. Mol. Sci. 2019, 20, 4342. [Google Scholar] [CrossRef] [Green Version]
- Prado, N.J.; Muñoz, E.M.; Farias Altamirano, L.E.; Aguiar, F.; Ponce Zumino, A.Z.; Sánchez, F.J.; Miatello, R.M.; Pueyo, E.; Diez, E.R. Reperfusion arrhythmias increase after superior cervical ganglionectomy due to conduction disorders and changes in repolarization. Int. J. Mol. Sci. 2020, 21, 1804. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkova, A.S.; Bernikova, O.G.; Mikhaleva, N.J.; Khramova, D.S.; Ovechkin, A.O.; Demidova, M.M.; Platonov, P.G.; Azarov, J.E. Melatonin prevents early but not delayed ventricular fibrillation in the experimental porcine model of acute ischemia. Int. J. Mol. Sci. 2020, 22, 328. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, functions and therapeutic benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.G.D.; Cipolla-Neto, J. A brief review about melatonin, a pineal hormone. Arch. Endocrinol. Metab. 2018, 62, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Won, E.; Na, K.S.; Kim, Y.K. Associations between melatonin, neuroinflammation and brain alterations in depression. Int. J. Mol. Sci. 2021, 23, 305. [Google Scholar] [CrossRef] [PubMed]
- Claustrat, B.; Brun, J.; Chazot, G. The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 2005, 9, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Reiter, R.J. An evolutionary view of melatonin synthesis and metabolism related to its biological functions in plants. J. Exp. Bot. 2020, 71, 4677–4689. [Google Scholar] [CrossRef]
- Tan, D.; Reiter, R.J. Mitochondria: The birth place, battle ground and the site of melatonin metabolism in cells. Melatonin Res. 2019, 2, 44–66. [Google Scholar] [CrossRef]
- Kim, T.K.; Lin, Z.; Tidwell, W.J.; Li, W.; Slominski, A.T. Melatonin and its metabolites accumulate in the human epidermis in vivo and inhibit proliferation and tyrosinase activity in epidermal melanocytes in vitro. Mol. Cell Endocrinol. 2015, 404, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV light touches the brain and endocrine system through skin, and why. Endocrinology 2018, 159, 1992–2007. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Pisarchik, A.; Semak, I.; Sweatman, T.; Wortsman, J. Characterization of the serotoninergic system in the C57BL/6 mouse skin. Eur. J. Biochem. 2003, 270, 3335–3344. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Wortsman, J.; Tobin, D.J. The cutaneous serotoninergic/melatoninergic system: Securing a place under the sun. FASEB J. 2005, 19, 176–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Kleszczyński, K.; Janjetovic, Z.; Sweatman, T.; Lin, Z.; Li, W.; Reiter, R.J.; Fischer, T.W.; Slominski, A.T. Metabolism of melatonin and biology activity of intermediates of melatoninergic pathway in human skin cells. FASEB J. 2013, 27, 2742–2755. [Google Scholar] [CrossRef] [PubMed]
- Iuvone, P.M.; Tosini, G.; Pozdeyev, N.; Haque, R.; Klein, D.C.; Chaurasia, S.S. Circadian clocks, clock networks, arylalkylamine N-acetyltransferase, and melatonin in the retina. Prog. Retin. Eye Res. 2005, 24, 433–456. [Google Scholar] [CrossRef] [PubMed]
- Hernàndex-González, O.; Herrera-Vargas, D.J.; Martinez-Leija, M.E.; Zavala-Reyes, D.; Portales-Pérez, D.P. The role of arylamine N-acetyltransferases in chronic degenerative diseasses: Their possible function in the immune system. BBA Mol. Cell Res. 2022, 1869, 119297. [Google Scholar]
- Liu, Y.J.; Zhuang, J.; Zhu, H.Y.; Shen, Y.X.; Tan, Z.L.; Zhou, J.N. Cultured rat cortical astrocytes synthesize melatonin: Absence of a diurnal rhythm. J. Pineal Res. 2007, 43, 232–238. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.K.; Kleszczyński, K.; Semak, I.; Janjetovic, Z.; Sweatman, T.; Skobowiat, C.; Steketee, J.D.; Lin, Z.; Postlethwaite, A.; et al. Characterization of serotonin and N-acetylserotonin system in human epidermis and skin cells. J. Pineal Res. 2020, 68, e12626. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.D.; Calcutt, M.W.; Malow, B.A.; Rose, K.L.; Hachey, D.L. Quantitation of melatonin and n-acetylserotonin in human plasma by nanoflow LC-MS/MS and electrospray LC-MS/MS. J. Mass Spectrom. 2012, 47, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Fisher, T.W.; Sweatman, T.W.; Semak, I.; Sayre, R.M.; Wortsman, J.; Slominski, A. Constitutive and UV-induced metabolism of melatonin in keratinocytes and cell-free system. FASEB J. 2006, 20, 1564–1566. [Google Scholar] [CrossRef] [Green Version]
- Bocheva, G.; Slominski, R.M.; Janjetovic, Z.; Kim, T.K.; Böhm, M.; Steinbrink, K.; Reiter, R.J.; Kleszczyński, K.; Slominski, A.T. Protective role of melatonin and its metabolites in skin aging. Int. J. Mol. Sci. 2022, 23, 1238. [Google Scholar] [CrossRef]
- Brzezinski, A.; Rai, S.; Purohit, A.; Pandi-Perumal, S.R. Melatonin, clock genes and mammalian reproduction: What is the link? Int. J. Mol. Sci. 2021, 22, 13240. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and microglia. Int. J. Mol. Sci. 2021, 22, 8296. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, Z.; Pan, S.; Zhang, H.; Fang, M.; Jiang, H.; Zhang, H.; Gao, Z.; Xu, K.; Li, Z.; et al. Melatonin protects against blood-brain barrier damage by inhibiting the TLR4/Nf-κB signalling pathway after LPS treatment in neonatal rats. Oncotarget 2017, 8, 31638–31654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alluri, H.; Wilson, R.L.; Anasooya Shaji, C.; Wiggins-Dohlvik, K.; Patel, S.; Liu, Y.; Peng, X.; Beeram, M.R.; Davis, M.L.; Huang, J.H.; et al. Melatonin preserves blood-brain barrier integrity and permeability via matrix metalloproteinase-9 inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qiu, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Akoi, K.; Zhao, K.; Yamazaki, F.; Sone, R.; Alvarez, G.E.; Kosiba, W.A.; Johnson, J.M. Exogenous melatonin administration modifies cutaneous vasoconstrictor response to whole body skin cooling in humans. J. Pineal Res. 2008, 44, 141–148. [Google Scholar] [CrossRef]
- Ray, C.A. Melatonin attenuates the sympathetic nerve responses to orthostatic stress in humans. J. Physiol. 2003, 551, 1043–1048. [Google Scholar] [CrossRef]
- Muler, M.D.; Sauder, C.L.; Ray, C.A. Melatonin attenuates the skin sympathetic nerve response to mental stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1382–H1386. [Google Scholar] [CrossRef] [Green Version]
- Fagali, N.; Catala, A. The antioxidant behaviour of melatonin and structural analogues during lipid peroxidation depends not only on their functional groups but also on the assay system. Biochem. Biophys. Res. Commun. 2012, 423, 873–877. [Google Scholar] [CrossRef]
- Wongprayoon, P.; Govitrapong, P. Melatonin receptor as a drug target for neuroprotection. Curr. Mol. Pharmacol. 2021, 4, 150–164. [Google Scholar] [CrossRef]
- Singhanat, K.; Apaijai, N.; Jaiwongkam, T.; Kerdphoo, S.; Chattipakron, S.C.; Chattipakron, N. Melatonin as a therapy in cardiac ischemia-reperfusion injury: Potential mechanisms by which MT2 activation mediates cardioprotection. J. Adv. Res. 2020, 29, 33–44. [Google Scholar] [CrossRef]
- Grossini, E.; Molinari, C.; Uberti, F.; Mary, D.A.; Vacca, G.; Caimmi, P.P. Intracoronary melatonin increases coronary blood flow and cardiac function through β-adrenoreceptors, MT1/MT2 receptors and nitric oxide in anesthetized pigs. J. Pineal Res. 2011, 51, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Naji, L.; Carrillo-Vico, A.; Guerrero, J.M.; Calvo, J.R. Expression of membrane and nuclear melatonin receptors in mouse peripheral organs. Life Sci. 2004, 74, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, B.; Angeloni, D.; Dominguez-Lopez, S.; Calderoni, S.; Mauro, A.; Fraschini, F.; Descarries, L.; Gobbi, G. Anatomical and cellular localization of melatonin MT1 and MT2 receptors in the adult rat brain. J. Pineal Res. 2015, 58, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Reiter, R.J.; Schlabritz-Loutsevitch, N.; Ostrom, R.S.; Slominski, A.T. Melatonin membrane receptors in peripheral tissues: Distribution and functions. Mol. Cell Endocrinol. 2012, 351, 152–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reppert, S.M.; Weaver, D.R.; Ebisawa, T. Cloning and characterization of mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron 1994, 13, 1177–1185. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.; Zisapel, N.; Cardinali, D.P. Physiological effect of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef]
- Dubocovich, M.L.; Markowska, M. Functional MT1 and MT2 melatonin receptors in mammals. Endocrine 2005, 27, 101–110. [Google Scholar] [CrossRef]
- Slominski, A.; Fischer, T.W.; Zmijewski, M.A.; Wortsman, J.; Semak, I.; Zbytek, B.; Slominski, R.M.; Tobin, D.J. On the role of melatonin in skin physiology and pathology. Endocrine 2005, 27, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.A.; Premont, R.T.; Lefkowitz, R.J. Heptahelical receptor signalling: Beyond the G protein paradigm. J. Cell Biol. 1999, 145, 927–932. [Google Scholar] [CrossRef]
- Nosjean, O.; Nicolas, J.P.; Klupsch, F.; Delagrange, P.; Canet, E.; Boutin, J.A. Comparative pharmacological studies of melatonin receptors: MT1, MT2 and MT3/QR2. Tissue distribution of MT3/QR2. Biochem. Pharmacol. 2001, 61, 1369–1379. [Google Scholar] [CrossRef]
- Slominski, A.T.; Semak, I.; Fischer, T.W.; Kim, T.K.; Kleszczyński, K.; Hardeland, R.; Reiter, R.J. Metabolism od melatonin in the skin: Why is it important? Exp. Dermatol. 2017, 26, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Fourtillan, J.B.; Brisson, A.M.; Gobin, P.; Ingrand, I.; Decourt, J.P.; Girault, J. Bioavailability of melatonin in humans after day-time administration of D7 melatonin. Biopharm. Drug Dispos. 2000, 21, 15–22. [Google Scholar] [CrossRef]
- Facciolá, G.; Hidestrand, M.; von Bahr, C.; Tybring, G. Cytochrome P450 isoforms involved in melatonin metabolism in human liver microsomes. Eur. J. Clin. Pharmacol. 2001, 56, 881–888. [Google Scholar] [CrossRef]
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes P450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; Srinivasan, V.; Maestroni, G.J.M.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin—Nature’s most versatile biological signal? FEBS J. 2006, 273, 2813–2838. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchaster, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Mayo, J.C.; Sainz, R.M.; Tan, D.X.; Hardeland, R.; Leon, J.; Rodriguez, C.; Reiter, R.J. Anti-inflammatory actions of melatonin and its metabolites, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK) in macrophages. J. Neuroimmunol. 2005, 165, 139–149. [Google Scholar] [CrossRef]
- Iwashita, H.; Matsumoto, Y.; Maruyama, Y.; Watanabe, K.; Chiba, A.; Hattori, A. The melatonin metabolite N1-acetyl-5-methoxykynuramine facilitates long-term object memory in young and aging mice. J. Pineal Res. 2021, 70, e12703. [Google Scholar] [CrossRef]
- De Almeida, E.A.; Martinez, G.R.; Klitzke, C.F.; de Medeiros, M.H.; Di Mascio, P. Oxidation of melatonin by singlet molecular oxygen (O2(1deltag)) produces N1-acetyl-N2-formyl-5-methoxykynurenine. J. Pineal Res. 2003, 35, 131–137. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Hardies, L.J.; Weintraub, S.T.; Shepherd, A.M. A novel melatonin metabolite, cyclic 3-hydroxymelatonin: A biomarker of in vivo hydroxyl radical generation. Biochem. Biophys. Res. Commun. 1998, 253, 614–620. [Google Scholar] [CrossRef]
- Florentino, S.A.; Bawany, M.H.; Ma, H.M. Acetylocholinesterase inhibitors to enhance recovery from traumatic brain injury: A comprehensive review and case reports. Brain Inj. 2022, 36, 441–454. [Google Scholar] [CrossRef]
- Cheung, R.T. The utility of melatonin in reducing cerebral damage resulting from ischemia and reperfusion. J. Pineal Res. 2003, 34, 153–160. [Google Scholar] [CrossRef]
- Wichniak, A.; Kania, A.; Siemiński, A.; Cubałą, W.J. Melatonin as a potential adjuvant treatment for COVID-19 beyond sleep disorders. Int. J. Mol. Sci. 2021, 22, 8623. [Google Scholar] [CrossRef]
- Paparrigopoulos, T.; Melissaki, A.; Tsekou, H.; Efthymiou, A.; Kribeni, G.; Baziotis, N.; Geronikola, X. Melatonin secretion after head injury: A pilot study. Brain Inj. 2006, 20, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Seifman, M.A.; Gomes, K.; Nguyen, P.N.; Bailey, M.; Rosenfeld, J.V.; Cooper, D.J.; Morganti-Kossmann, M.C. Measurement of serum melatonin in intensive care unit patients: Changes in traumatic brain injury, trauma and medical conditions. Front. Neurol. 2014, 5, 237. [Google Scholar] [CrossRef] [Green Version]
- Lorente, L.; Martín, M.M.; Abreu-González, P.; Pérez-Cejas, A.; Ramos, L.; Argueso, M.; Solé-Violán, J.; Cáceres, J.J.; Jiménez, A.; Garcia-Marin, V. Serum melatonin levels in survivors and non-survivors patients with traumatic brain injury. BMC Neurol. 2017, 17, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumaier, F.; Weiss, M.; Veldeman, M.; Kotliar, K.; Wiesmann, M.; Schulze-Steinen, H.; Höllig, A.; Clusmann, H.; Schubert, G.A.; Albanna, W. Changes in endogenous daytime melatonin levels after aneurysmal subarachnoid haemorrhage—Preliminary findings from an observational cohort study. Clin. Neurol. Neurosurg. 2021, 208, 106870. [Google Scholar] [CrossRef] [PubMed]
- Melville, K.I.; Blum, B.; Shister, H.E.; Silver, M.D. Cardiac ischemic changes and arrhythmias induced by hypothalamic stimulation. Am. J. Cardiol. 1963, 12, 781–791. [Google Scholar] [CrossRef]
- Whitnall, M.H. Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog. Neurobiol. 1993, 40, 573–629. [Google Scholar] [CrossRef]
- Torres-Farfan, C.; Richter, H.G.; Rojas-García, P.; Vergara, M.; Forcelledo, M.L.; Valladares, L.E.; Torrealba, F.; Valenzuela, G.J. Seroín-Ferreí; M mt1 Melatonin receptor in the primate adrenal; gland: Inhibition of adrenocorticotropic-stimulated cortisol production by melatonin. J. Clin. Endocrinol. Metab. 2003, 88, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Salman, M.; Kaushik, P.; Tabassum, H.; Parvez, S. Melatonin provides neuroprotection following traumatic brain injury-promoted mitochondrial perturbation in Wistar rat. Cell Mol. Neurobiol. 2021, 41, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuated traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Lee, S.H.; Oh, D.S.; Kim, Y.U. Melatonin inhibits neuronal dysfunction-associates with neuroinflammation by atopic psychological stress in NC/Nga atopic-like mouse models. J. Pineal Res. 2017, 63, e12420. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Wang, B.; Wang, L.; Abraham, N.; Tao, K.; Huang, L.; Shi, W.; Dong, Y.; Qu, Y. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibitinh endoplasmatic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J. Pineal Res. 2017, 62, e12395. [Google Scholar] [CrossRef]
- Yan, E.B.; Frugier, T.; Lim, C.K.; Heng, B.; Sundaram, G.; Tan, M.; Rosenfeld, J.V.; Walker, D.W.; Guillemin, G.J.; Morganti-Kossmann, M.C. Activation of the kynurenine pathway and increase production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J. Neuroinflamm. 2015, 12, 110. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.; Pires, A.S.; Tan, V.; Babu Chidambaram, S.; Guillemin, G.J. Effect of sleep deprivation on the tryptophan metabolism. Int. J. Trypt. Res. 2020, 23, 1178646920970902. [Google Scholar] [CrossRef]
- Millischer, V.; Heinzl, M.; Faka, A.; Resl, M.; Trepci, A.; Klammer, C.; Egger, M.; Dieplinger, B.; Clodi, M.; Schwieler, L. Intravenous administration of LPS activates the kynurenine pathway in healthy male human subjests: A prospective placebo-controlled cross-over trial. J. Neuroinflamm. 2021, 18, 158. [Google Scholar] [CrossRef]
- Zhang, Z.; Rasmussen, L.; Saraswati, M.; Koehler, R.; Robertson, C.; Kannan, S. Traumatic injury leads to inflammation and altered tryptophan metabolism in the Juvenile rabbit brain. J. Neurotrauma 2018, 11, 74–86. [Google Scholar] [CrossRef]
- Miner, S.E.; Pahal, D.; Nichols, L.; Darwood, A.; Nield, L.E.; Wulffhart, Z. Sleep disruption is associated with increased ectopy and cardiac arrest in hospitalized adults. Sleep 2016, 39, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Cakici, M.; Dogan, A.; Cetin, M.; Suner, A.; Caner, A.; Polat, M.; Kaya, H.; Abus, S.; Akturk, E. Negative effect of acute sleep deprivation on left ventricular functions and cardiac repolarization in healthy young adults. Pacing Clin. Electrophysiol. 2015, 38, 713–722. [Google Scholar] [CrossRef]
- Dabrowski, W.; Schlegel, T.T.; Wosko, J.; Rola, R.; Rzecki, Z.; Malbrain, M.L.; Jaroszynski, A. Changes in spatial QRS-T angle and QTc interval in patients with traumatic brain injury with or without intra-abdominal hypertension. J. Electrocardiol. 2018, 51, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Canno, W.B. Voodoo death. Am. Anthropol. 1942, 44, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.G. The exciting causes of ventricular fibrillation in animals under chloroform anesthesia. Heart 1913, 4, 1912–1913. [Google Scholar]
- Ng, G.A.; Mantravadi, R.; Walker, W.H.; Ortin, W.G.; Choi, B.R.; de Groat, W.; Salama, G. Sympathetic nerve stimulation produces spatial heterogeneities of action potential restitution. Heart Rhytm. 2009, 6, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Katona, P.G. Biomedical engineering in heart-brain medicine: A review. Clevel. Clin. J. Med. 2010, 77 (Suppl. S3), S46–S50. [Google Scholar] [CrossRef] [PubMed]
- Silvani, A.; Calandra-Buonaura, G.; Dampney, R.A.L.; Cortelli, P. Brain-heart interactions: Physiology and clinical implications. Phil. Trans. R. Soc. A. 2016, 374, 20150181. [Google Scholar] [CrossRef] [PubMed]
- Hadaya, J.; Ardell, J.L. Autonomic modulation for cardiovascular disease. Front. Physiol. 2020, 11, 617459. [Google Scholar] [CrossRef] [PubMed]
- Verrier, R.L.; Clavert, A.; Lown, B. Effect of posterior hypothalamic stimulation on ventricular fibrillation threshold. Am. J. Physiol. 1975, 228, 923–927. [Google Scholar] [CrossRef]
- Oppenheimer, S.M. Neurogenic cardiac effects of cerebrovascular disease. Curr. Opin. Neurol. 1994, 7, 20–24. [Google Scholar] [CrossRef]
- Seifert, F.; Kallmünzer, B.; Gutjahr, I.; Breuer, L.; Winder, K.; Kaschka, I.; Kloska, S.; Doerfler, A.; Hilz, M.J.; Schwab, S.; et al. Neuroanatomical correlates of severe cardiac arrhythmias in acute ischemic stroke. J. Neurol. 2015, 262, 1182–1190. [Google Scholar] [CrossRef]
- Ay, H.; Koroshetz, W.J.; Benner, T.; Vangel, M.G.; Melinosky, C.; Arsava, E.M.; Ayata, C.; Zhu, M.; Schwamm, L.H.; Sorensen, A.G. Neuroanatomic correlates of stroke-related myocardial injury. Neurology 2006, 66, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Infanger, D.W.; Cao, X.; Butler, S.D.; Burmeister, M.A.; Zhou, Y.; Stupinski, J.A.; Sharma, R.V.; Davisson, R.L. Silencing nox4 in the paraventricular nucleus improves myocardial infarction-induced cardiac dysfunction by attenuating sympathoexcitation and periinfarct apoptosis. Circ. Res. 2010, 106, 1763–1774. [Google Scholar] [CrossRef]
- Reuss, S.; Olcese, J.; Vollrath, L. Electrical stimulation of the hypothalamic paraventricular nuclei inhibits pineal melatonin synthesis in male rats. Neuroendocrinology 1985, 41, 192–196. [Google Scholar] [CrossRef]
- Yang, J.B.; Kang, Y.M.; Zhang, C.; Yu, X.J.; Chen, W.S. Infusion of melatonin into paraventricular nucleus ameliorates myocardial ischemia-reperfusion injury by regulating oxidative stress and inflammatory cytokines. J. Cardiovasc. Pharmacol. 2019, 74, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sato, K.; Yamamoto, S.I.; Matsuyama, N.; Shimohama, T.; Matsunaga, A.; Obuchi, S.; Shiba, Y.; Shimizu, S.; Izumi, T. Sympathetic nervous activity and myocardial damage immediately after subarachnoid hemorrhage in unique animal model. Stroke 2002, 33, 1671–1676. [Google Scholar] [CrossRef] [PubMed]
- Mazzeo, A.T.; Micalizzi, A.; Mascia, L.; Scicolone, A.; Siracusano, L. Brain-heart crosstalk: The many faces of stress-related cardiomyopathy syndromes in anaesthesia and intensive care. Br. J. Anaesth. 2014, 112, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Novityzky, D.; Wicomb, W.N.; Cooper, D.K.C.; Rose, A.G.; Reichart, B. Prevention of myocardial injury during brain death by total cardiac sympathectomy in the Chacma baboon. Ann. Thorac. Surg. 1986, 41, 520–524. [Google Scholar] [CrossRef]
- Lyon, A.R.; Rees, P.S.; Prasad, S.; Poole-Wilson, P.A.; Harding, S.E. Stress (Takotsubo) cardiomyopathy: A novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 22–29. [Google Scholar] [CrossRef]
- De Aquino, L.V.; Dos Santos, R.V.T.; Antunes, H.K.M.; Behn, C.; Viscor, G.; Lira, F.S.; Bittar, I.G.L.; Caris, A.V.; Tufik, S.; De Mello, M.T. Melatonin and sleep responses to normobaric hypoxia and aerobic physical exercise: A randomized controlled trial. Physiol. Behav. 2018, 196, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Ben Dhia, I.; Maaloul, R.; Marzougui, H.; Ghroubi, S.; Kallel, C.; Driss, T.; Elleuch, M.H.; Ayadi, F.; Turki, M.; Hammouda, O. Melatonin reduces muscle damage, inflammation and oxidative stress induced by exhaustive exercise in people with overweight/obesity. Physiol. Int. 2022, 109, 78–89. [Google Scholar] [CrossRef]
- Jou, M.J.; Peng, T.; Yu, P.Z.; Jou, S.B.; Reiter, R.J.; Chen, J.Y. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J. Pineal Res. 2007, 43, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Yürüker, V.; Naziroğlu, M.; Senol, N. Reduction in traumatic brain injury-induced oxidative stress, apoptosis and calcium entry in rat hippocampus by melatonin: Possible involvement of TRPM2 channels. Metab. Brain Dis. 2015, 30, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Cai, E.L.; Yang, C.; Ye, C.Y.; Zeng, P.; Wang, X.M.; Fang, Y.Y.; Cheng, Z.K.; Wang, Q.; Cao, F.Y.; et al. Protection of melatonin against acidosis-induced neuronal injuries. J. Cell Mol. Med. 2020, 24, 6928–6942. [Google Scholar] [CrossRef]
- Ortiz-Franco, M.; Planells, E.; Quintero, B.; Acuña-Castroviejo, D.; Rusanova, I.; Escames, G.; Molina-López, J. Effect of melatonin supplementation on antioxidant status and DNA damage in high intensity trained athletes. Int. J. Sports Med. 2017, 38, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Ghafourifar, P.; Richter, C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997, 418, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Korge, P.; Ping, P.; Weiss, J.N. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: Modulation by nitric oxide. Circ. Res. 2008, 103, 873–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano, B.M.; Colin-Gonzalez, A.L.; Santamaria, A.; Tunez, I. The role of melatonin in multiple scletorir, Huntington’s disease and cerebral ischemia. CNS Neurol. Disord. Drug Targets 2014, 13, 1096–1119. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Burkhardt, S.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Shohami, E.; Huo, Y.S.; Hardeland, R.; Reioter, R.J. N1-acetyl-N2-formyl-5-methoxykynuramine, a biogenic amine and melatonin metabolite, functions as a potent antioxidant. FASEB J. 2001, 15, 2294–2296. [Google Scholar] [CrossRef]
- Ressmeyer, A.R.; Mayo, J.C.; Zelosko, V.; Sainz, R.M.; Tan, D.X.; Poeggeler, B.; Antolin, I.; Zsizsik, B.K.; Reiter, R.J.; Hardeland, R. Antioxidant properties of melatonin metabolite N1-acetyl-5-methoxykynuramine (AMK): Scavenging of free radicals and prevention of protein destruction. Redox Rep. 2003, 8, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Burkhardt, S.; Reiter, R.J.; Tan, D.X.; Hardeland, R.; Cabrera, J.; Karbownik, M. DNA oxidatively damaged by chromium III and H2O2 is protected by the antioxidants melatonin, N1-acetyl-N2-formyl-5-methoxykynuramine, resverator and uric acid. Int. J. Biochem. Cell Biol. 2001, 33, 775–783. [Google Scholar] [CrossRef]
- León, J.; Escames, G.; Rodríguez, M.I.; López, L.C.; Tapias, V.; Entrena, A.; Camacho, E.; Carrión, M.D.; Gallo, M.A.; Espinosa, A.; et al. Inhibition of neuronal nitric oxide synthase activity by N1-acetyl-5-methoxykynuramine, a brain metabolite of melatonin. J. Neurochoem. 2006, 98, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S. Ca+2/calmodulin and presynaptic short-term plasticity. ISRN Neurol. 2011, 2011, 919043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilal, S.; Chai, Y.L.; Ikram, M.K.; Elangovan, S.; Yeow, T.B.; Xin, X.; Chong, J.Y.; Venketasubramanian, N.; Richards, A.M.; Chong, J.P.; et al. Markers of cardiac dysfunction in cognitive impairment and dementia. Medicine 2015, 91, e297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, B.; Guo, X.; Wang, X.; Guo, M.; Wang, Z.; Xue, T.; Li, H.; Xu, T.; Ye, S.; et al. Brain-heart interactions underlying traditional Tibetan Buddhist meditation. Cereb. Cortex 2020, 30, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Niijima, A.; Chun, S.J.; Shima, T.; Bizot-Espiard, J.G.; Guardiola-Lemaitre, B.; Nagai, K. Effect of intravenous administration of melatonin on the efferent activity of the adrenal nerve. J. Auton. Nerv. Syst. 1998, 71, 134–138. [Google Scholar] [CrossRef]
- Fernandes, P.A.; Tamura, E.K.; D’Argenio-Garcia, L.; Muxel, S.M.; da Silveira Cruz-Machado, S.; Marcola, M.; Carvalho-Sousa, C.E.; Cecon, E.; Ferreira, Z.S.; Markus, R.P. Dual effect of catecholamine and corticosterone crosstals on pineal gland melatonin synthesis. Neuroendocrinology 2017, 104, 126–134. [Google Scholar] [CrossRef]
- Mortani-Barbosa, E.J.; Ferreira, Z.S.; Marckus, R.P. Purinergic and noradrenergic cotransmission in the rat pineal gland. Eur. J. Pharmacol. 2000, 401, 59–62. [Google Scholar] [CrossRef]
- Sugama, S.; Takenouchi, T.; Hashimoto, M.; Ohata, H.; Takenaka, Y.; Kakinuma, Y. Stress-induced microglial activation occurs through β-adrenergic receptor: Noradrenaline as a key neurotransmitter in microglial activation. J. Neuroinflamm. 2019, 16, 266. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Wang, Y.Y.; Chang, C.Y.; Wu, C.C.; Chen, W.Y.; Kuan, Y.H.; Liao, S.L.; Chen, C.J. Effects of β-adrenergic blockade on metabolic and inflammatory responses in a rat model of ischemic stroke. Cells 2020, 9, 1373. [Google Scholar] [CrossRef]
- Birketvedt, G.S.; Sundsfjord, J.; Florholmen, J.R. Hypothalamic-pituitary-adrenal axis in the might eating syndrome. Am. J. Physiol. Endocrin. Metab. 2002, 282, E366–E369. [Google Scholar] [CrossRef] [Green Version]
- Weidenfeld, Y.; Schmidt, U.; Nir, I. The effect of exogenous melatonin on the hypothalamic-pituitary-adrenal axis in intact and pinealectomized rats under basal and stress conditions. J. Pineal Res. 1993, 14, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Seifman, M.A.; Adamides, A.A.; Nguyen, P.N.; Vallance, S.A.; Cooper, D.J.; Kossmann, T.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Endogenous melatonin increases in cerebrospinal fluid of patients after severe traumatic brain injury and correlates with oxidative and metabolic disarray. J. Cereb. Blood Flow Metab. 2008, 28, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Arroyo-Ucar, E.; Avanzas, P.; Reiter, R.J. Global left ventricular longitudinal strain is associated with decreased melatonin levels in patients with acute myocardial infarction: A two-dimensional speckle tracking study. Biomarkers 2013, 18, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Arroyo-Ucar, E.; Reiter, R.J. Decreased level of melatonin in serum predicts left ventricular remodeling after acute myocardial infarction. J. Pineal Res. 2012, 53, 319–323. [Google Scholar] [CrossRef]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Jose, M.; Consuegra-Sanchez, L.; Piccolo, R.; Gonzalez-Gonzalez, J.; Garcia-Camarero, T.; del Mar Garcia-Saiz, M.; Aldea-Perona, A.; Reiter, R.J.; et al. Usefulness of early treatment with melatonin to reduce infarct size in patients with ST-segment elevation myocardial infarction receiving percutaneous coronary intervention (from the melatonin adjunct in the acute myocardial infarction treated with angioplasty trial). Am. J. Cardiol. 2017, 120, 522–526. [Google Scholar]
- Scheer, F.A.; Van Montfrans, G.A.; van Someren, E.J.; Mairuhu, G.; Buijs, R.M. Daily nighttime melatonin reduces blood pressure in male patients with essential hypertension. Hypertension 2004, 43, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Grossman, E.; Laudon, M.; Yalcin, R.; Zengil, H.; Peleg, E.; Sharabi, Y.; Kamari, Y.; Shen-Orr, Z.; Zisapel, N. Melatonin reduces night blood pressure in patients with nocturnal hypertension. Am. J. Med. 2006, 119, 898–902. [Google Scholar] [CrossRef]
- Hicks, J.J.; Montes-Cortes, D.H.; Cruz-Dominguez, M.P.; Medina-Santillan, R.; Olivares-Corichi, I.M. Antioxidants decrease reperfusion induced arrhythmias in myocardial infarction with ST-elevation. Front. Biosci. 2007, 12, 2029. [Google Scholar] [CrossRef] [Green Version]
- Sallinen, P.; Mänttäri, S.; Leskinen, H.; Ilves, M.; Vakkuri, O.; Ruskoaho, H.; Saarela, S. The effect of myocardial infarction on synthesis, concentration and receptor expression of endogenous melatonin. J. Pineal Res. 2007, 42, 254–260. [Google Scholar] [CrossRef]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Gracia, M.J.; Sanchez, J.; Marrero, F.; de Armas-Trujillo, D. Decreased nocturnal melatonin levels during acute myocardial infarction. J. Pineal Res. 2002, 33, 248–252. [Google Scholar] [CrossRef]
- Dwaich, K.H.; Al-Amar, F.G.Y.; Al-Sheibani, B.I.M.; Al-Aubaidy, H.A. Melatonin effects on myocardial ischemia-reperfusion injury: Impact on the outcome in patients undergoing coronary artery bypass grafting surgery. Int. J. Cardiol. 2016, 221, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Z.; Chua, C.C.; Ma, Y.S.; Youngberg, G.A.; Hamdy, R.; Chua, B.H. Melatonin as an effective protector against doxorubicin-induced cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H254–H263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubar, R.; Setoodeh, M.; Savari, Z.F.; Ahmadi, S.; Karami, A.; Khademi, S. The effect of melatonin on the serum level of interleukin 6 and interleukin 9 in coronary artery bypass grafting surgery. Asian J. Anesthesiol. 2020, 58, 35–44. [Google Scholar]
- Deng, W.G.; Tang, S.T.; Tseng, H.P.; Wu, K.K. Melatonin supplesses macrophage cyclooxygenase-2 and inducible notric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524. [Google Scholar] [CrossRef]
- Benitex-King, G. Melatonin as a cytoskeletal modulator: Implication for cell physiology and disease. J. Pineal Res. 2006, 40, 1–9. [Google Scholar] [CrossRef]
- Sedova, K.A.; Bernikova, O.G.; Cuprova, J.I.; Ivanova, A.D.; Kutaeva, G.A.; Pliss, M.G.; Lopatina, E.V.; Vaykshnorayte, M.A.; Diez, E.R.; Azarov, J.E. Association between antiarrhythmic, electrophysiological and antioxidant effect of melatonin in ischemia/reperfusion. Int. J. Mol. Sci. 2019, 20, 6331. [Google Scholar] [CrossRef] [Green Version]
- Diez, E.R.; Prados, L.V.; Carrion, A.; Ponce, Z.A.Z.; Miatello, R.M. A novel electrophysiologic effect of melatonin on ischemia/reperfusion-induced arrhythmias in isolated rat hearts. J. Pieal Res. 2009, 46, 155–160. [Google Scholar] [CrossRef]
- Mashaly, H.A.; Provenico, J.J. Inflammation as a link between brain injury and heart damage: The model of subarachnoid hemorrhage. Cleve Clin. J. Med. 2008, 75 (Suppl. S2), S26–S30. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Yan, T.; Li, L.; Chopp, M.; Venkat, P.; Qian, Y.; Li, R.; Wu, R.; Li, W.; Lu, M.; et al. Immune response mediates cardiac dysfunction after traumatic brain injury. J. Neurotrauma 2019, 36, 619–629. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekała, A.; Płotek, W.; Siwicka-Gieroba, D.; Sołek-Pastuszka, J.; Bohatyrewicz, R.; Biernawska, J.; Kotfis, K.; Bielacz, M.; Jaroszyński, A.; Dabrowski, W. Melatonin and the Brain–Heart Crosstalk in Neurocritically Ill Patients—From Molecular Action to Clinical Practice. Int. J. Mol. Sci. 2022, 23, 7094. https://doi.org/10.3390/ijms23137094
Bekała A, Płotek W, Siwicka-Gieroba D, Sołek-Pastuszka J, Bohatyrewicz R, Biernawska J, Kotfis K, Bielacz M, Jaroszyński A, Dabrowski W. Melatonin and the Brain–Heart Crosstalk in Neurocritically Ill Patients—From Molecular Action to Clinical Practice. International Journal of Molecular Sciences. 2022; 23(13):7094. https://doi.org/10.3390/ijms23137094
Chicago/Turabian StyleBekała, Artur, Włodzimierz Płotek, Dorota Siwicka-Gieroba, Joanna Sołek-Pastuszka, Romuald Bohatyrewicz, Jowita Biernawska, Katarzyna Kotfis, Magdalena Bielacz, Andrzej Jaroszyński, and Wojciech Dabrowski. 2022. "Melatonin and the Brain–Heart Crosstalk in Neurocritically Ill Patients—From Molecular Action to Clinical Practice" International Journal of Molecular Sciences 23, no. 13: 7094. https://doi.org/10.3390/ijms23137094