
Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease

 ,
,  ,
,  ,
,  , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
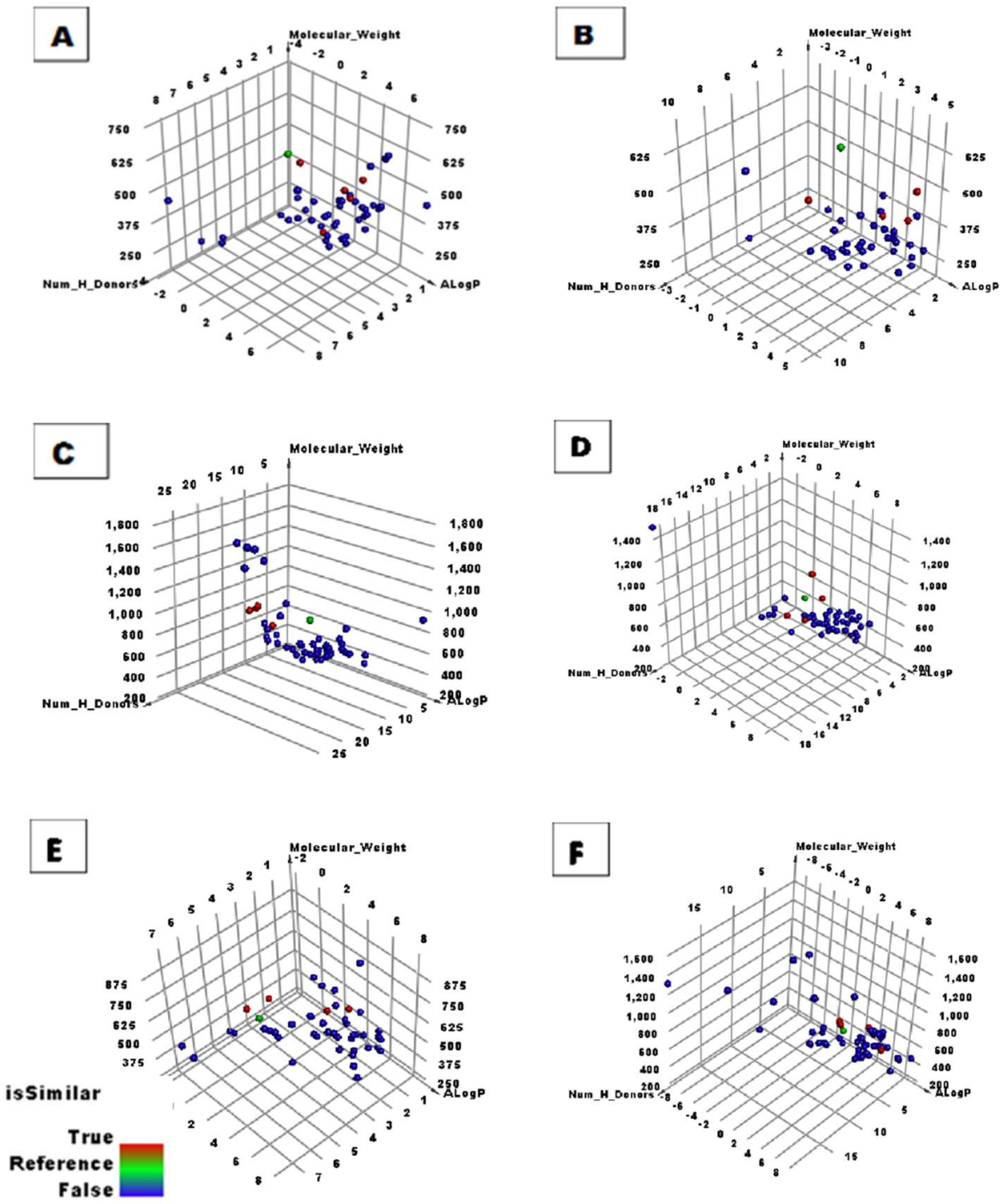
2.1. Structural Similarity Detection
2.2. Structural Fingerprint Study
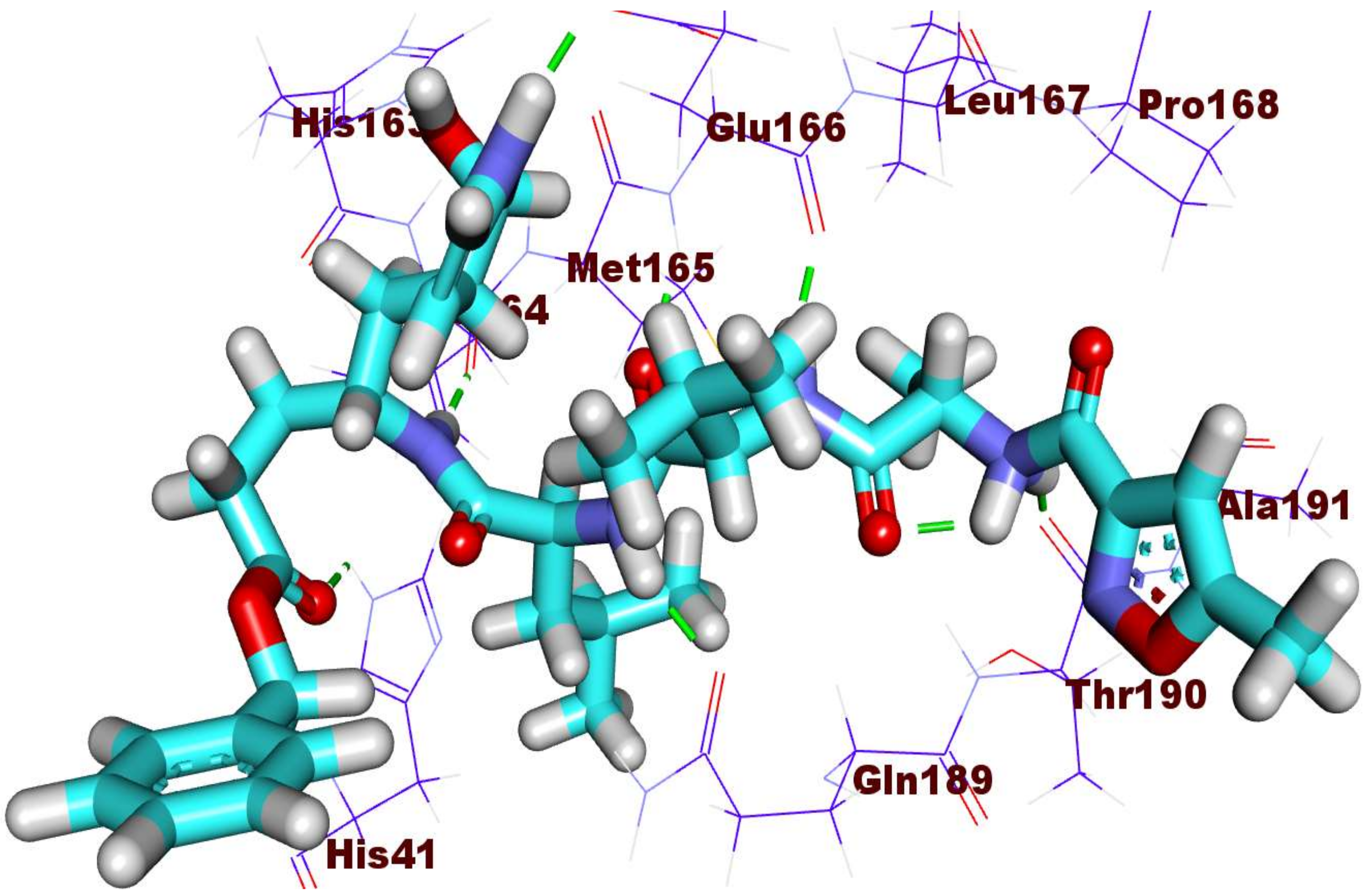
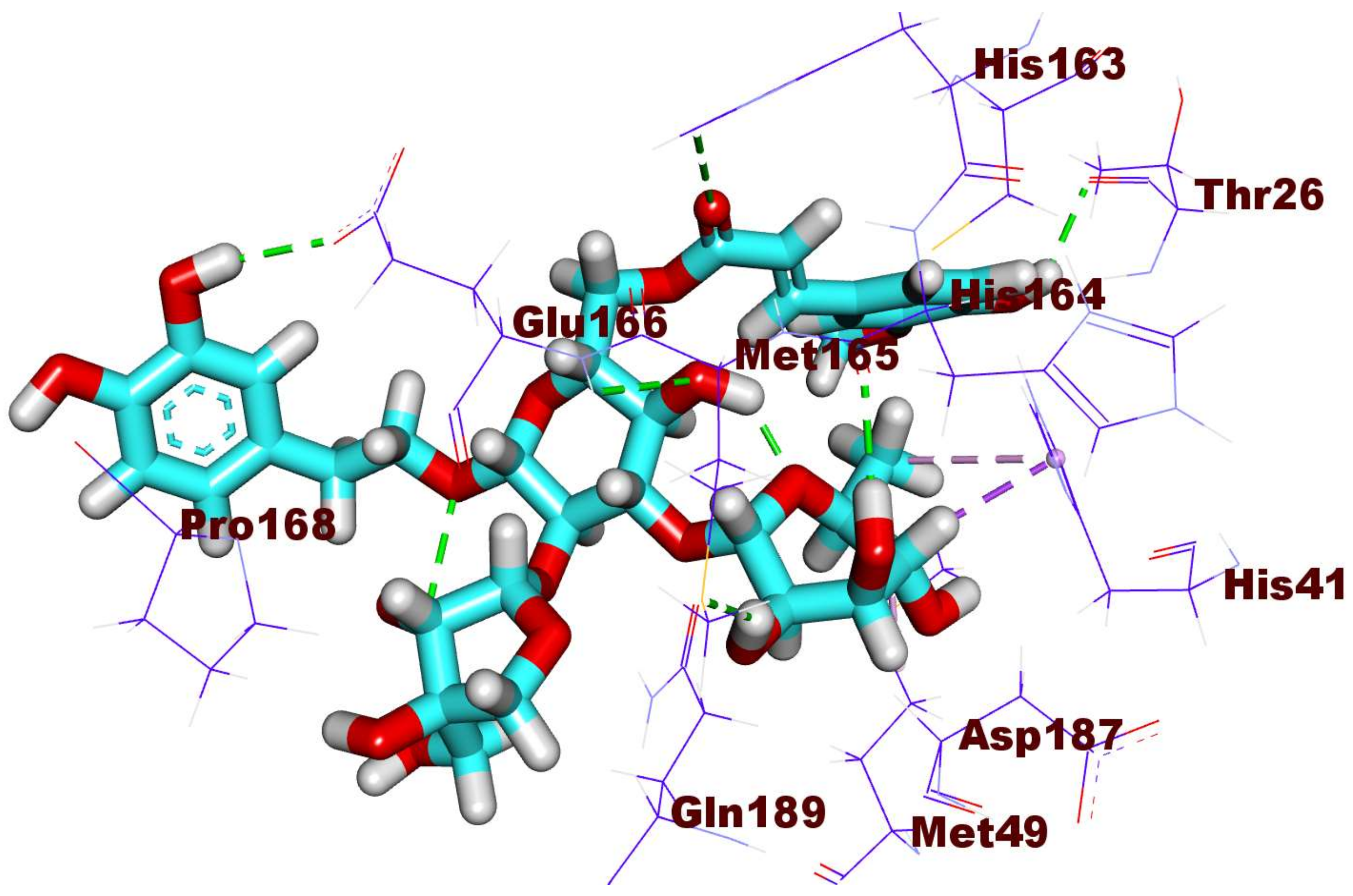
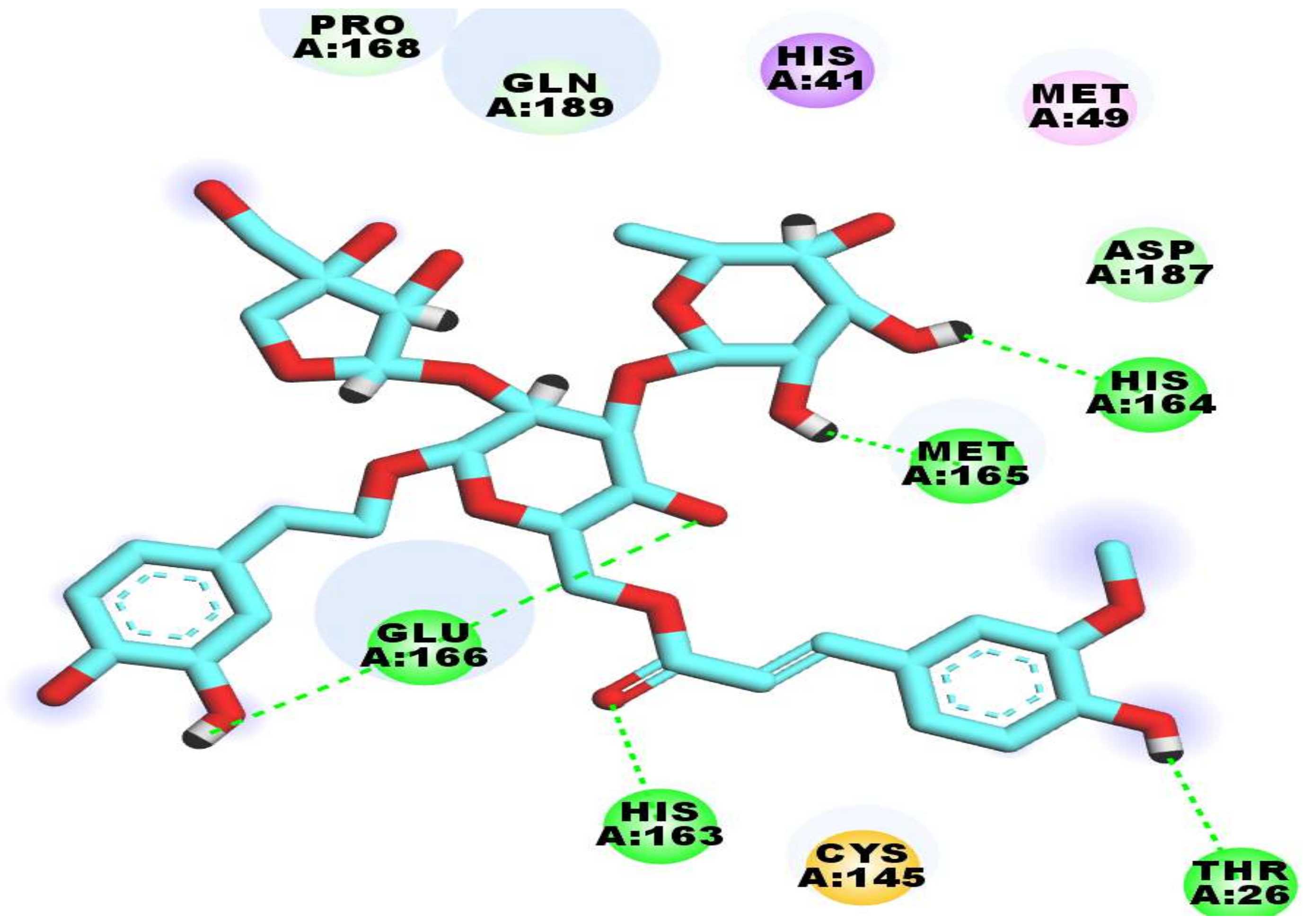
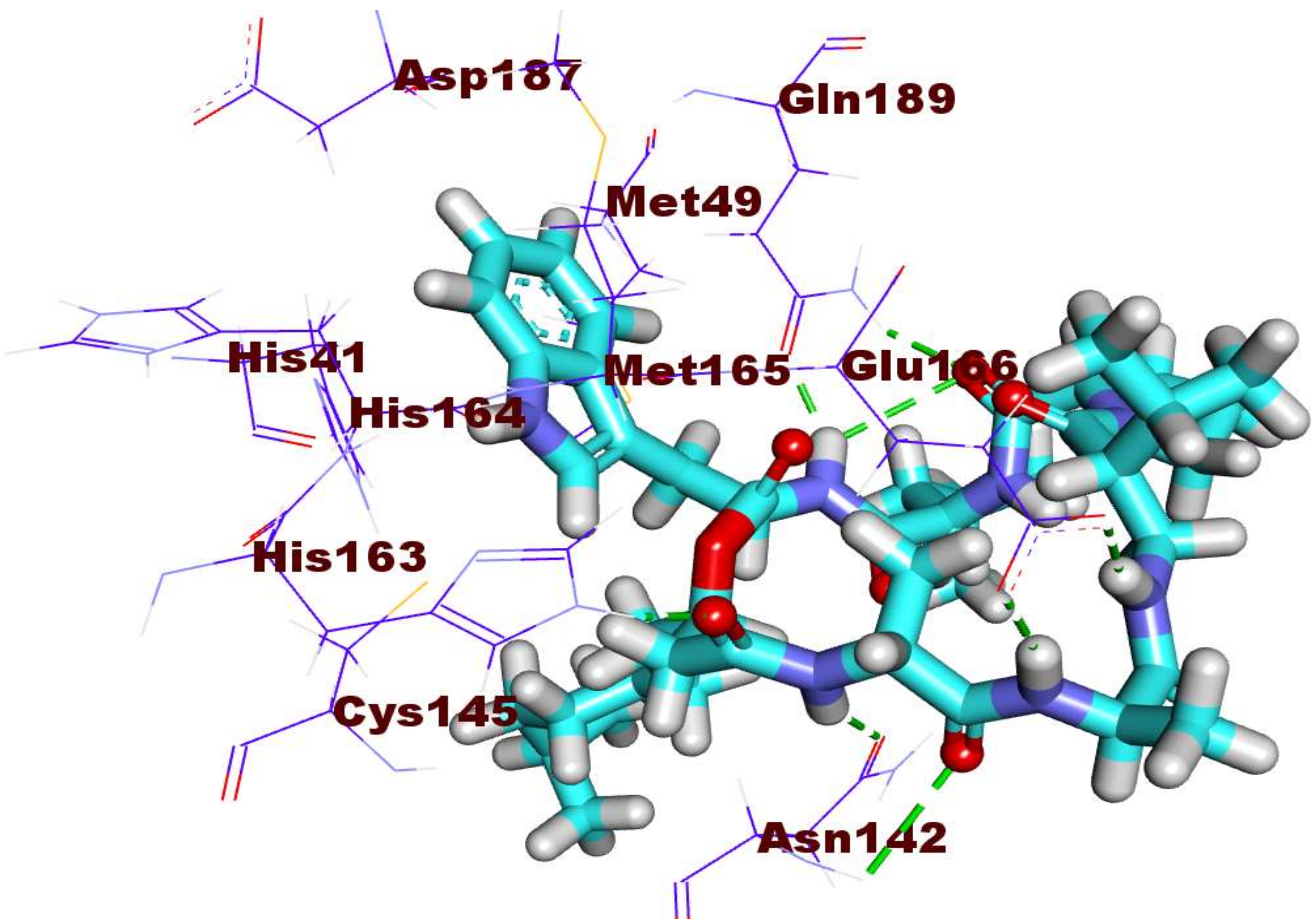
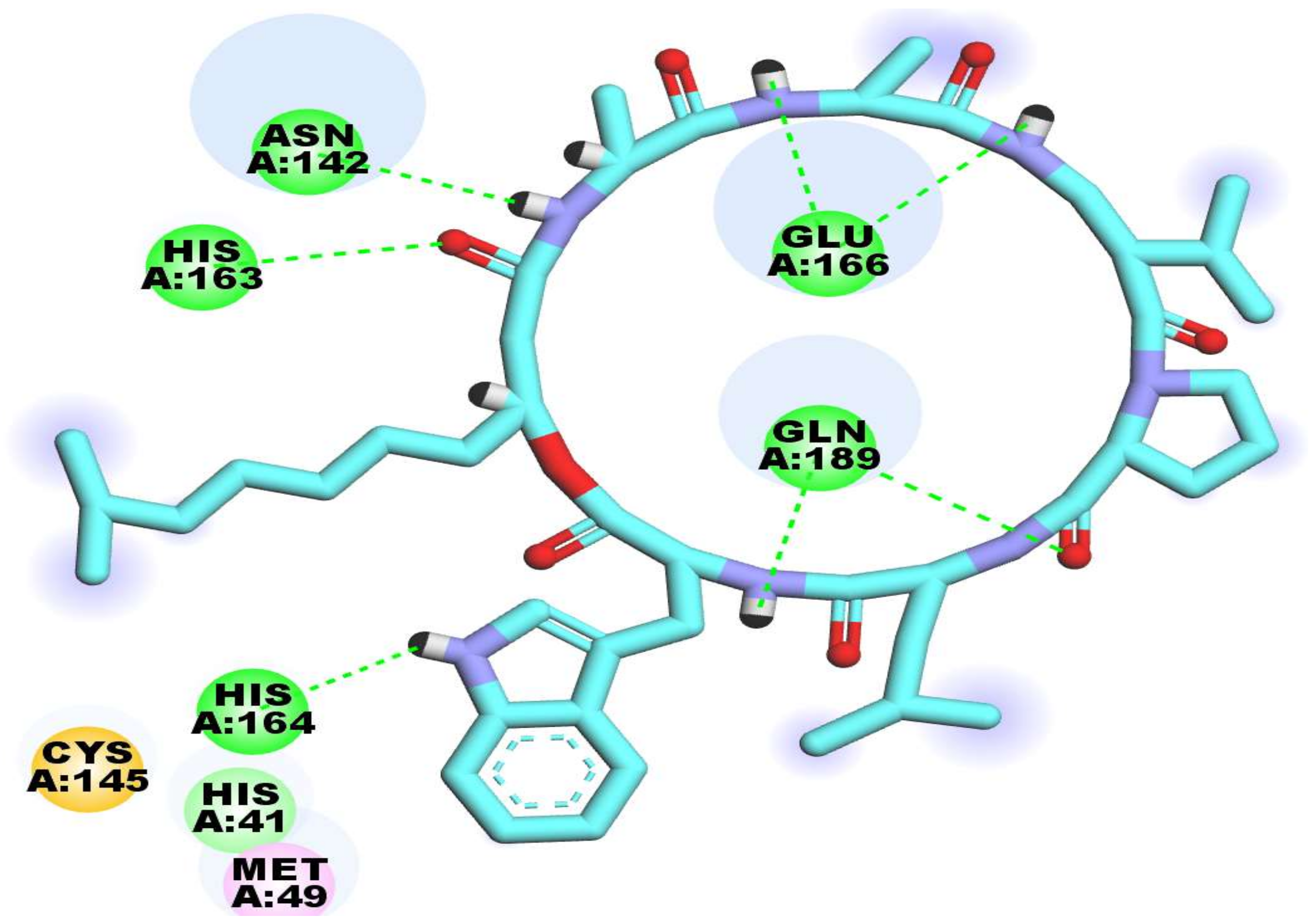
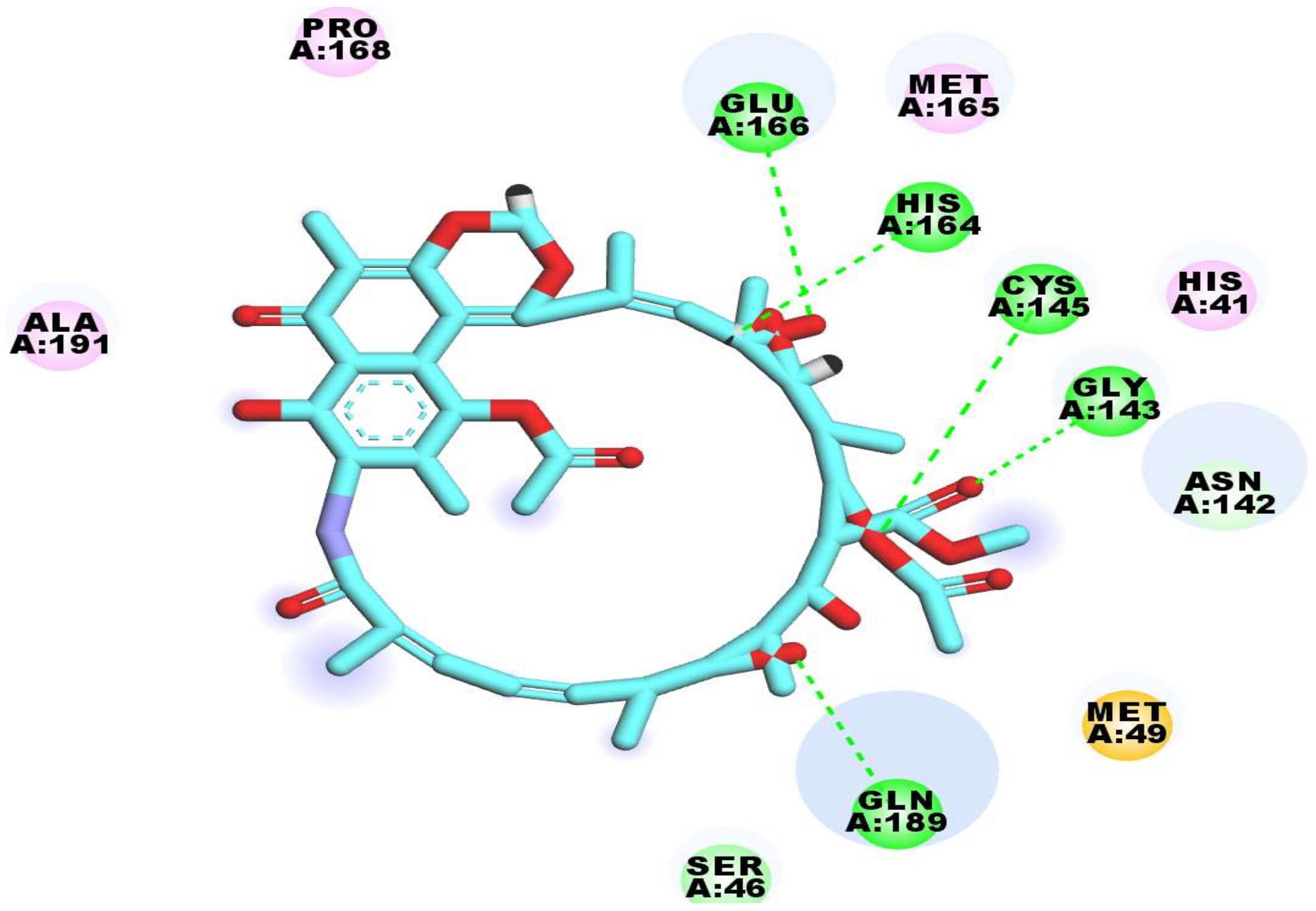
2.3. Docking Studies
2.4. Toxicity Models
2.5. Molecular Dynamics
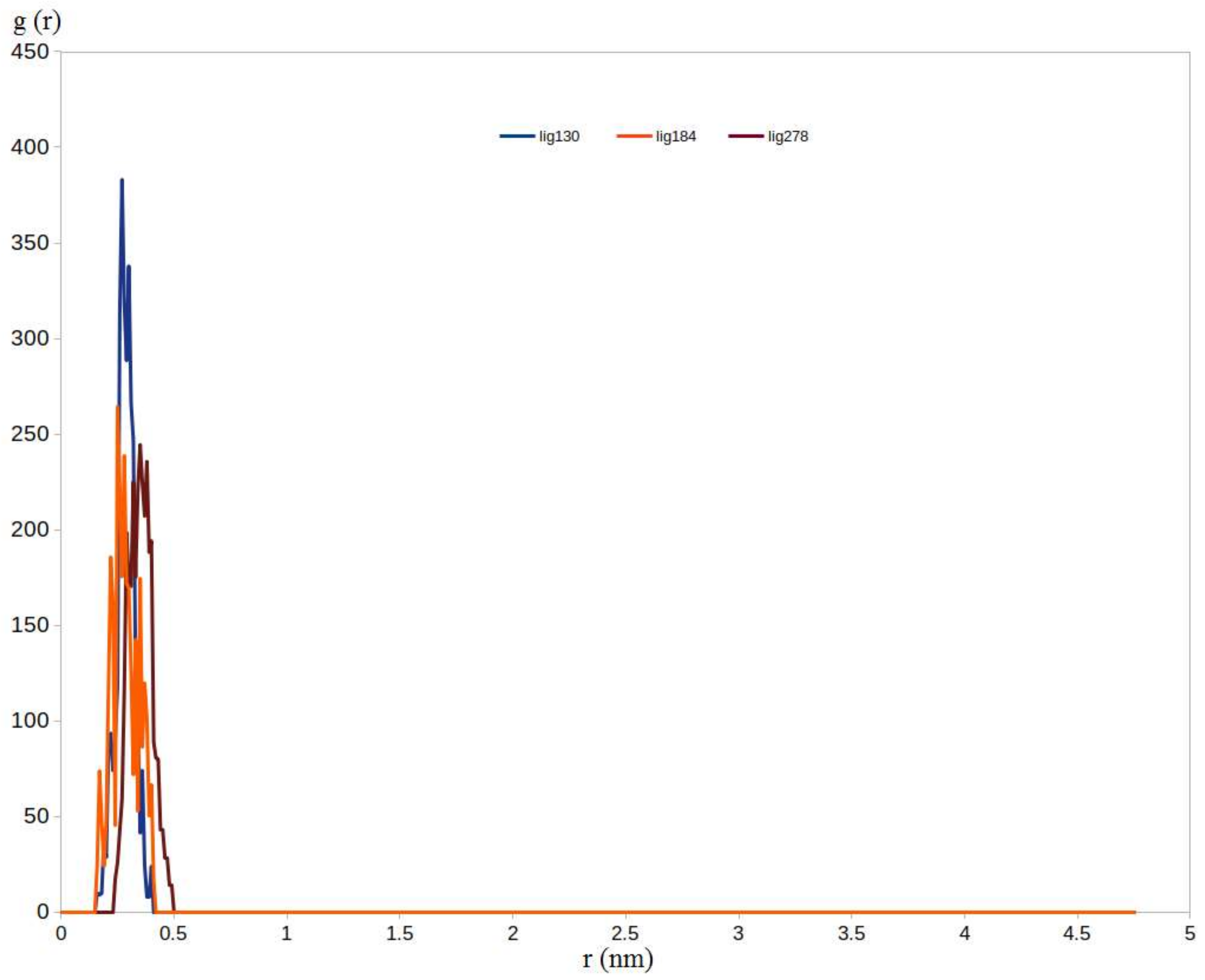
2.5.1. RMSD, RMSF, and RDF Analysis
2.5.2. Binding Free Energy Calculations Using MM-PBSA Approach
3. Method
3.1. Molecular Similarity Detection
3.2. Molecular Fingerprint Detection
3.3. Docking Studies
3.4. Toxicity Studies
3.5. MDS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 9 February 2022).
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Elwan, A.; Elkady, H. Discovery of new VEGFR-2 inhibitors based on bis ([1, 2, 4] triazolo)[4, 3-a: 3′, 4′-c] quinoxaline derivatives as anticancer agents and apoptosis inducers. J. Enzyme Inhib. Med. Chem. 2021, 36, 1093–1114. [Google Scholar] [CrossRef] [PubMed]
- Belal, A. 3D-Pharmacophore Modeling, Molecular Docking, and Virtual Screening for Discovery of Novel CDK4/6 Selective Inhibitors. Russ. J. Bioorg. Chem. 2021, 47, 317–333. [Google Scholar] [CrossRef]
- Belal, A. Pyrrolizines as Potential Anticancer Agents: Design, Synthesis, Caspase-3 activation and Micronucleus (MN) Induction. Anti-Cancer Agents Med. Chem. 2018, 18, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Marrone, T.J.; Briggs, A.J.M.; McCammon, J.A. Structure-based drug design: Computational advances. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 71–90. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wang, Y.; Li, W.; Li, H.; Yang, L.; Wang, J.; Mahdy, H.A.; Mehany, A.; Jaiash, D.A.; Santali, E.Y. Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem. 2020, 2020, 1631243. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Fares, M.; Al-Rashood, S.T.; Al-Rashood, K.A.; Abdel-Aziz, M.M.; Soliman, D.H. Synthesis, biological evaluation and 2D-QSAR study of halophenyl bis-hydrazones as antimicrobial and antitubercular agents. Int. J. Mol. Sci. 2015, 16, 8719–8743. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Hadni, H.; Elhallaoui, M. 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J. Chem. 2020, 44, 6553–6565. [Google Scholar] [CrossRef]
- Yadav, D.K.; Khan, F.; Negi, A.S. Pharmacophore modeling, molecular docking, QSAR, and in silico ADMET studies of gallic acid derivatives for immunomodulatory activity. J. Mol. Modeling 2012, 18, 2513–2525. [Google Scholar] [CrossRef]
- Belal, A.; Elanany, M.A.; Santali, E.Y.; Al-Karmalawy, A.A.; Aboelez, M.O.; Amin, A.H.; Abdellattif, M.H.; Mehany, A.B.; Elkady, H. Screening a Panel of Topical Ophthalmic Medications against MMP-2 and MMP-9 to Investigate Their Potential in Keratoconus Management. Molecules 2022, 27, 3584. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Dahab, M.A.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Mahdy, H.A.; Elkady, H. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: Design, molecular modeling, and synthesis. Bioorg. Chem. 2021, 110, 104807. [Google Scholar] [CrossRef]
- El-Adl, K.; Ibrahim, M.-K.; Alesawy, M.S.; Eissa, I.H. [1, 2, 4] Triazolo [4, 3-c] quinazoline and bis ([1, 2, 4] triazolo)[4, 3-a: 4′, 3′-c] quinazoline derived DNA intercalators: Design, synthesis, in silico ADMET profile, molecular docking and anti-proliferative evaluation studies. Bioorg. Med. Chem. 2021, 30, 115958. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. [Google Scholar] [CrossRef]
- Zhang, W.; Pei, J.; Lai, L. Computational multitarget drug design. J. Chem. Inf. Modeling 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Youssef, M.I.; Zhou, Y.; Eissa, I.H.; Wang, Y.; Zhang, J.; Jiang, L.; Hu, W.; Qi, J.; Chen, Z. Tetradecyl 2, 3-dihydroxybenzoate alleviates oligodendrocyte damage following chronic cerebral hypoperfusion through IGF-1 receptor. Neurochem. Int. 2020, 138, 104749. [Google Scholar] [CrossRef]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg. Chem. 2021, 112, 104947. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Eissa, I.H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Elkady, H.; Elwan, A. Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers. J. Enzyme Inhib. Med. Chem. 2021, 36, 1760–1782. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Alaa, E.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Taghour, M.S.; Eissa, I.H. Discovery of new 3-methylquinoxalines as potential anti-cancer agents and apoptosis inducers targeting VEGFR-2: Design, synthesis, and in silico studies. J. Enzyme Inhib. Med. Chem. 2021, 36, 1732–1750. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Metwaly, A.M.; Ghoneim, M.M.; Eissa, I.H.; Elsehemy, I.A.; Mostafa, A.E.; Hegazy, M.M.; Afifi, W.M.; Dou, D. Traditional ancient Egyptian medicine: A review. Saudi J. Biol. Sci. 2021, 28, 5823–5832. [Google Scholar] [CrossRef]
- Han, X.; Yang, Y.; Metwaly, A.M.; Xue, Y.; Shi, Y.; Dou, D. The Chinese herbal formulae (Yitangkang) exerts an antidiabetic effect through the regulation of substance metabolism and energy metabolism in type 2 diabetic rats. J. Ethnopharmacol. 2019, 239, 111942. [Google Scholar] [CrossRef] [PubMed]
- Suleimen, Y.M.; Metwaly, A.M.; Mostafa, A.E.; Elkaeed, E.B.; Liu, H.-W.; Basnet, B.B.; Suleimen, R.N.; Ishmuratova, M.Y.; Turdybekov, K.M.; Van Hecke, K. Isolation, Crystal Structure, and In Silico Aromatase Inhibition Activity of Ergosta-5, 22-dien-3β-ol from the Fungus Gyromitra esculenta. J. Chem. 2021, 2021, 5529786. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Lianlian, Z.; Luqi, H.; Deqiang, D. Black ginseng and its saponins: Preparation, phytochemistry and pharmacological effects. Molecules 2019, 24, 1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metwaly, A.M.; Wanas, A.S.; Radwan, M.M.; Ross, S.A.; ElSohly, M.A. New α-Pyrone derivatives from the endophytic fungus Embellisia sp. Med. Chem. Res. 2017, 26, 1796–1800. [Google Scholar] [CrossRef]
- Yassin, A.M.; El-Deeb, N.M.; Metwaly, A.M.; El Fawal, G.F.; Radwan, M.M.; Hafez, E.E. Induction of apoptosis in human cancer cells through extrinsic and intrinsic pathways by Balanites aegyptiaca furostanol saponins and saponin-coated silvernanoparticles. Appl. Biochem. Biotechnol. 2017, 182, 1675–1693. [Google Scholar] [CrossRef]
- Metwaly, A.M.; Fronczek, F.R.; Ma, G.; Kadry, H.A.; Atef, A.; Mohammad, A.-E.I.; Cutler, S.J.; Ross, S.A. Antileukemic α-pyrone derivatives from the endophytic fungus Alternaria phragmospora. Tetrahedron Lett. 2014, 55, 3478–3481. [Google Scholar] [CrossRef] [Green Version]
- Imieje, V.O.; Zaki, A.A.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Ali, Z.; Khan, I.A.; Falodun, A. Antileishmanial Derivatives of Humulene from Asteriscus hierochunticus with in silico Tubulin Inhibition Potential. Rec. Nat. Prod. 2021, 150–171. [Google Scholar]
- Metwaly, A.M.; Ghoneim, M.M.; Musa, A. Two new antileishmanial diketopiperazine alkaloids from the endophytic fungus Trichosporum sp. Derpharmachemica 2015, 7, 322. [Google Scholar]
- Jalmakhanbetova, R.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M.; Suleimen, Y.M. Synthesis and Molecular Docking of Some Grossgemin Amino Derivatives as Tubulin Inhibitors Targeting Colchicine Binding Site. J. Chem. 2021, 2021, 5586515. [Google Scholar] [CrossRef]
- Zhanzhaxina, A.; Suleimen, Y.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Suleimen, R.; Ishmuratova, M.; Akatan, K.; Luyten, W. In vitro and in silico cytotoxic and antibacterial activities of a diterpene from cousinia alata schrenk. J. Chem. 2021, 2021, 5542455. [Google Scholar] [CrossRef]
- Sharaf, M.H.; El-Sherbiny, G.M.; Moghannem, S.A.; Abdelmonem, M.; Elsehemy, I.A.; Metwaly, A.M.; Kalaba, M.H. New combination approaches to combat methicillin-resistant Staphylococcus aureus (MRSA). Sci. Rep. 2021, 11, 4240. [Google Scholar] [CrossRef]
- Liu, L.; Luo, S.; Yu, M.; Metwaly, A.M.; Ran, X.; Ma, C.; Dou, D.; Cai, D. Chemical Constituents of Tagetes patula and Their Neuroprotecting Action. Nat. Prod. Commun. 2020, 15, 1934578X20974507. [Google Scholar]
- Wang, Y.-M.; Ran, X.-K.; Riaz, M.; Yu, M.; Cai, Q.; Dou, D.-Q.; Metwaly, A.M.; Kang, T.-G.; Cai, D.-C. Chemical constituents of stems and leaves of Tagetespatula L. and its fingerprint. Molecules 2019, 24, 3911. [Google Scholar] [CrossRef] [Green Version]
- Metwaly, A.M.; Kadry, H.A.; Atef, A.; Mohammad, A.-E.I.; Ma, G.; Cutler, S.J.; Ross, S.A. Nigrosphaerin A a new isochromene derivative from the endophytic fungus Nigrospora sphaerica. Phytochem. Lett. 2014, 7, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.; Metwaly, A.M. Ligand and structure-based in silico determination of the most promising SARS-CoV-2 nsp16-nsp10 2′-o-Methyltransferase complex inhibitors among 3009 FDA approved drugs. Molecules 2022, 27, 2287. [Google Scholar] [CrossRef]
- Imieje, V.O.; Zaki, A.A.; Metwaly, A.M.; Mostafa, A.E.; Elkaeed, E.B.; Falodun, A. Comprehensive In Silico Screening of the Antiviral Potentialities of a New Humulene Glucoside from Asteriscus hierochunticus against SARS-CoV-2. J. Chem. 2021, 2021, 5541876. [Google Scholar] [CrossRef]
- Agbowuro, A.A.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018, 38, 1295–1331. [Google Scholar] [CrossRef]
- Du, Q.-S.; Wang, S.-Q.; Zhu, Y.; Wei, D.-Q.; Guo, H.; Sirois, S.; Chou, K.-C. Polyprotein cleavage mechanism of SARS CoV Mpro and chemical modification of the octapeptide. Peptides 2004, 25, 1857–1864. [Google Scholar] [CrossRef]
- Hegyi, A.; Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002, 83, 595–599. [Google Scholar] [CrossRef]
- Liang, P.-H. Characterization and inhibition of SARS-coronavirus main protease. Curr. Top. Med. Chem. 2006, 6, 361–376. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Alesawy, M.S.; Abdallah, A.E.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M. In Silico Studies of Some Isoflavonoids as Potential Candidates against COVID-19 Targeting Human ACE2 (hACE2) and Viral Main Protease (Mpro). Molecules 2021, 26, 2806. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; El-Aziz, A.; Mohamed, T.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Khalifa, M.M.; Elkaeed, E.B.; Hafez, E.E.; Alsfouk, A.A.; Metwaly, A.M. In Silico Exploration of Potential Natural Inhibitors against SARS-Cov-2 nsp10. Molecules 2021, 26, 6151. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I. In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-Like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules 2021, 26, 6593. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry: Miniperspective. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef]
- Altamash, T.; Amhamed, A.; Aparicio, S.; Atilhan, M. Effect of hydrogen bond donors and acceptors on CO2 absorption by deep eutectic solvents. Processes 2020, 8, 1533. [Google Scholar] [CrossRef]
- Wan, Y.; Tian, Y.; Wang, W.; Gu, S.; Ju, X.; Liu, G. In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches. RSC Adv. 2018, 8, 40529–40543. [Google Scholar] [CrossRef] [Green Version]
- Turchi, M.; Cai, Q.; Lian, G. An evaluation of in-silico methods for predicting solute partition in multiphase complex fluids–A case study of octanol/water partition coefficient. Chem. Eng. Sci. 2019, 197, 150–158. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Enoch, S.J.; Ezendam, J.; Sewald, K.; Roggen, E.L.; Cochrane, S. An adverse outcome pathway for sensitization of the respiratory tract by low-molecular-weight chemicals: Building evidence to support the utility of in vitro and in silico methods in a regulatory context. Appl. Vitr. Toxicol. 2017, 3, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ren, J.-X.; Ma, J.-X.; Ding, L. Development of an in silico prediction model for chemical-induced urinary tract toxicity by using naïve Bayes classifier. Mol. Divers. 2019, 23, 381–392. [Google Scholar] [CrossRef]
- Escamilla-Gutiérrez, A.; Ribas-Aparicio, R.M.; Córdova-Espinoza, M.G.; Castelán-Vega, J.A. In silico strategies for modeling RNA aptamers and predicting binding sites of their molecular targets. Nucleosides Nucleotides Nucleic Acids 2021, 40, 798–807. [Google Scholar] [CrossRef]
- Kaushik, A.C.; Kumar, A.; Bharadwaj, S.; Chaudhary, R.; Sahi, S. Ligand-Based Approach for In-silico Drug Designing. In Bioinformatics Techniques for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2018; pp. 11–19. [Google Scholar]
- Sheridan, R.P.; Kearsley, S.K. Why do we need so many chemical similarity search methods? Drug Discov. Today 2002, 7, 903–911. [Google Scholar] [CrossRef]
- Bender, A.; Jenkins, J.; Scheiber, J.; Sukuru, S.C.K.; Glick, M.; Davies, J.W. How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef]
- Moreira, D.D.L.; Leitão, G.G. Quantitative determination of liriodenine and moupinamide in five species of Mollinedia by high performance liquid chromatography. Phytochem. Anal. 2001, 12, 223–225. [Google Scholar] [CrossRef]
- Shukla, R.; Singh, S.; Singh, A.; Misra, K. Two pronged approach for prevention and therapy of COVID-19 (SARS-CoV-2) by a multi-targeted herbal drug, a component of ayurvedic decoction. Eur. J. Integr. Med. 2021, 43, 101268. [Google Scholar] [CrossRef]
- Hiranuma, S.; Shibata, M.; Hudlicky, T. Studies in cephalotaxus alkaloids. Stereospecific total synthesis of homoharringtonine. J. Org. Chem. 1983, 48, 5321–5326. [Google Scholar] [CrossRef]
- Choy, K.-T.; Wong, A.Y.-L.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.-H.; Huang, X. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Mallik, A.K.; Talapatra, B. Pongaglabol, a new hydroxyfuranoflavone, and aurantiamide acetate, a dipeptide from the flowers of Pongamia glabra. Phytochemistry 1980, 19, 1199–1202. [Google Scholar] [CrossRef]
- Yoon, C.-S.; Kim, D.-C.; Lee, D.-S.; Kim, K.-S.; Ko, W.; Sohn, J.H.; Yim, J.H.; Kim, Y.-C.; Oh, H. Anti-neuroinflammatory effect of aurantiamide acetate from the marine fungus Aspergillus sp. SF-5921: Inhibition of NF-κB and MAPK pathways in lipopolysaccharide-induced mouse BV2 microglial cells. Int. Immunopharmacol. 2014, 23, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, Z.; Feng, Q.; Liang, X.; Li, J.; Zanin, M.; Jiang, Z.; Zhong, N. Aurantiamide acetate from Baphicacanthus cusia root exhibits anti-inflammatory and anti-viral effects via inhibition of the NF-κB signaling pathway in Influenza A virus-infected cells. J. Ethnopharmacol. 2017, 199, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Sahu, N.; Mahato, S.B. Production of the alkaloids emetine and cephaeline in callus cultures of Cephaelis ipecacuanha. Planta Med. 1988, 54, 504–506. [Google Scholar] [CrossRef]
- Kumar, R.; Afsar, M.; Khandelwal, N.; Chander, Y.; Riyesh, T.; Dedar, R.K.; Gulati, B.R.; Pal, Y.; Barua, S.; Tripathi, B.N. Emetine suppresses SARS-CoV-2 replication by inhibiting interaction of viral mRNA with eIF4E. Antivir. Res. 2021, 189, 105056. [Google Scholar] [CrossRef]
- Itoh, A.; Ikuta, Y.; Baba, Y.; Tanahashi, T.; Nagakura, N. Ipecac alkaloids from Cephaelis acuminata. Phytochemistry 1999, 52, 1169–1176. [Google Scholar] [CrossRef]
- Alrasheid, A.A.; Babiker, M.Y.; Awad, T.A. Evaluation of certain medicinal plants compounds as new potential inhibitors of novel corona virus (COVID-19) using molecular docking analysis. Silico Pharmacol. 2021, 9, 10. [Google Scholar] [CrossRef]
- El Sayed, K.A. Natural products as antiviral agents. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2000; Volume 24, pp. 473–572. [Google Scholar]
- Lee, I.-A.; Joh, E.-H.; Kim, D.-H. Arctigenin isolated from the seeds of Arctium lappa ameliorates memory deficits in mice. Planta Med. 2011, 77, 1525–1527. [Google Scholar] [CrossRef]
- Fu, L.; Xu, P.; Liu, N.; Yang, Z.; Zhang, F.; Hu, Y. Antiviral effect of Arctigenin compound on influenza virus. Tradit. Chin. Drug Res. Clin. Pharmacol. 2008, 4, 1–4. [Google Scholar]
- Shen, Y.-F.; Liu, L.; Chen, W.-C.; Hu, Y.; Zhu, B.; Wang, G.-X. Evaluation on the antiviral activity of arctigenin against spring viraemia of carp virus. Aquaculture 2018, 483, 252–262. [Google Scholar] [CrossRef]
- Páska, C.; Innocenti, G.; Kunvári, M.; László, M.; Szilágyi, L. Lignan production by Ipomoea cairica callus cultures. Phytochemistry 1999, 52, 879–883. [Google Scholar] [CrossRef]
- Qian, X.-J.; Jin, Y.-S.; Chen, H.-S.; Xu, Q.-Q.; Ren, H.; Zhu, S.-Y.; Tang, H.-L.; Wang, Y.; Zhao, P.; Qi, Z.-T. Trachelogenin, a novel inhibitor of hepatitis C virus entry through CD81. J. Gen. Virol. 2016, 97, 1134–1144. [Google Scholar] [CrossRef]
- Kernan, M.R.; Sendl, A.; Chen, J.L.; Jolad, S.D.; Blanc, P.; Murphy, J.T.; Stoddart, C.A.; Nanakorn, W.; Balick, M.J.; Rozhon, E.J. Two new lignans with activity against influenza virus from the medicinal plant Rhinacanthus nasutus. J. Nat. Prod. 1997, 60, 635–637. [Google Scholar] [CrossRef]
- Kernan, M.R.; Amarquaye, A.; Chen, J.L.; Chan, J.; Sesin, D.F.; Parkinson, N.; Ye, Z.-J.; Barrett, M.; Bales, C.; Stoddart, C.A. Antiviral phenylpropanoid glycosides from the medicinal plant Markhamia lutea. J. Nat. Prod. 1998, 61, 564–570. [Google Scholar] [CrossRef]
- Dimitrova, P.; Alipieva, K.; Stojanov, K.; Milanova, V.; Georgiev, M.I. Plant-derived verbascoside and isoverbascoside regulate Toll-like receptor 2 and 4-driven neutrophils priming and activation. Phytomedicine 2019, 55, 105–118. [Google Scholar] [CrossRef]
- Martins, F.O.; Esteves, P.F.; Mendes, G.S.; Barbi, N.S.; Menezes, F.S.; Romanos, M.T. Verbascoside isolated from Lepechinia speciosa has inhibitory activity against HSV-1 and HSV-2 in vitro. Nat. Prod. Commun. 2009, 4, 1934578X0900401217. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Fujioka, S.; Nakanishi, K.; Koreeda, M.; Kurokawa, T. Extraction of ponasterone a and ecdysterone from podocarpaceae and related plants. Steroids 1967, 10, 557–565. [Google Scholar] [CrossRef]
- Kimura, N.; Kainuma, M.; Inoue, T.; Chan, E.; Tangah, J.; Baba, K.; Oshiro, N.; Okamoto, C. Botany, uses, chemistry and bioactivities of mangrove plants V: Acrostichum aureum and A. speciosum. ISME/GLOMIS Electron. J. 2017, 15, 1–6. [Google Scholar]
- Unwalla, H.J.; Li, M.-J.; Kim, J.D.; Li, H.T.; Ehsani, A.; Alluin, J.; Rossi, J.J. Negative feedback inhibition of HIV-1 by TAT-inducible expression of siRNA. Nat. Biotechnol. 2004, 22, 1573–1578. [Google Scholar] [CrossRef]
- Pugach, P.; Ray, N.; Klasse, P.J.; Ketas, T.J.; Michael, E.; Doms, R.W.; Lee, B.; Moore, J.P. Inefficient entry of vicriviroc-resistant HIV-1 via the inhibitor-CCR5 complex at low cell surface CCR5 densities. Virology 2009, 387, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Bandara, B.R.; Jayasinghe, L.; Karunaratne, V.; Wannigama, G.P.; Bokel, M.; Kraus, W.; Sotheeswaran, S. Ecdysterone from stem of Diploclisia glaucescens. Phytochemistry 1989, 28, 1073–1075. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Gloer, J.B.; Cook, J.C., Jr.; Mizsak, S.A.; Scahill, T.A. Structures of the didemnins, antiviral and cytotoxic depsipeptides from a Caribbean tunicate. J. Am. Chem. Soc. 1981, 103, 1857–1859. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Gloer, J.B.; Hughes, R.G.; Renis, H.E.; McGovren, J.P.; Swynenberg, E.B.; Stringfellow, D.A.; Kuentzel, S.L.; Li, L.H. Didemnins: Antiviral and antitumor depsipeptides from a Caribbean tunicate. Science 1981, 212, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Otto, C.S.; Scheuer, P.J.; Dunbar, D.C. Kahalalides: Bioactive peptides from a marine mollusk Elysiarufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996, 61, 6594–6600. [Google Scholar] [CrossRef]
- Gustafson, K.R.; Oku, N.; Milanowski, D.J. Antiviral marine natural products. Curr. Med. Chem. Anti-Infect. Agents 2004, 3, 233–249. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.; Pannell, L.K. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus Mycale. J. Am. Chem. Soc. 1988, 110, 4850–4851. [Google Scholar] [CrossRef]
- Chou, K.-C.; Wei, D.-Q.; Zhong, W.-Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003, 308, 148–151. [Google Scholar] [CrossRef]
- Sagar, S.; Kaur, M.; Minneman, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef] [Green Version]
- Ichiba, T.; Yoshida, W.Y.; Scheuer, P.J.; Higa, T.; Gravalos, D.G. Hennoxazoles, bioactive bisoxazoles from a marine sponge. J. Am. Chem. Soc. 1991, 113, 3173–3174. [Google Scholar] [CrossRef]
- Groweiss, A.; Look, S.A.; Fenical, W. Solenolides, new antiinflammatory and antiviral diterpenoids from a marine octocoral of the genus Solenopodium. J. Org. Chem. 1988, 53, 2401–2406. [Google Scholar] [CrossRef]
- Shivanika, C.; Kumar, D.; Ragunathan, V.; Tiwari, P.; Sumitha, A. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020, 1, 585–611. [Google Scholar]
- Blunt, J.; Hartshorn, M.; McLennan, T.; Munro, M.; Robinson, W.T.; Yorke, S. Thyrsiferol: A squalene-derived metabolite of Laurencia thyrsifera. Tetrahedron Lett. 1978, 19, 69–72. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Shield, L.S.; Cohen-Parsons, M. Antiviral Substances. In Pharmaceutical and Bioactive Natural Products; Attaway, D.H., Zaborsky, O.R., Eds.; Springer: Boston, MA, USA, 1993; pp. 309–342. [Google Scholar]
- Palma, F.B.; Araújo, M.; Urones, J.; Barcala, P.; Marcos, I.; Ligtow, A.; Gravalos, M. Antiviral Activity of Metabolites from the Brown Seaweed Cystoseira usneoides. Planta Med. 1991, 57, A19. [Google Scholar] [CrossRef]
- Urones, J.; Araujo, M.; Palma, F.B.; Basabe, P.; Marcos, I.; Moro, R.; Lithgow, A.; Pineda, J. Meroterpenes from Cystoseira usneoides II. Phytochemistry 1992, 31, 2105–2109. [Google Scholar] [CrossRef]
- Baker, B.J.; Okuda, R.K.; Yu, P.T.; Scheuer, P.J. Punaglandins: Halogenated antitumor eicosanoids from the octocoral Telesto riisei. J. Am. Chem. Soc. 1985, 107, 2976–2977. [Google Scholar] [CrossRef]
- Mahalaxmi, Y.; Sathish, T.; Rao, C.S.; Prakasham, R. Corn husk as a novel substrate for the production of rifamycin B by isolated Amycolatopsis sp. RSP 3 under SSF. Process Biochem. 2010, 45, 47–53. [Google Scholar] [CrossRef]
- Becker, Y. Antiviral agents from natural sources. Pharmacol. Ther. 1980, 10, 119–159. [Google Scholar] [CrossRef]
- Milavetz, B.I.; Carter, W.A. Streptovaricins. Pharmacol. Ther. Part A 1977, 1, 289–305. [Google Scholar] [CrossRef]
- Arcamone, F.; Penco, S.; Orezzi, P.; Nicolella, V.; Pirelli, A. Structure and synthesis of distamycin A. Nature 1964, 203, 1064–1065. [Google Scholar] [CrossRef]
- Grunicke, H.; Puschendorf, B.; Werchau, H. Mechanism of action of distamycin A and other antibiotics with antiviral activity. Rev. Physiol. Biochem. Pharmacol. 1976, 75, 69–96. [Google Scholar]
- Broyles, S.S.; Kremer, M.; Knutson, B.A. Antiviral activity of distamycin A against vaccinia virus is the result of inhibition of postreplicative mRNA synthesis. J. Virol. 2004, 78, 2137–2141. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; He, Q.-X.; Wang, J.; Hu, Y.; Wang, Y.-Q.; Lin, Z.-H. In silico design of novel benzohydroxamate-based compounds as inhibitors of histone deacetylase 6 based on 3D-QSAR, molecular docking, and molecular dynamics simulations. New J. Chem. 2020, 44, 21201–21210. [Google Scholar] [CrossRef]
- Ieritano, C.; Campbell, J.L.; Hopkins, W.S. Predicting differential ion mobility behaviour in silico using machine learning. Analyst 2021, 146, 4737–4743. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Ali, M.; Rashid, U.; Imran, S.; Uddin, N.; Khan, K.M. Molecular hybridization conceded exceptionally potent quinolinyl-oxadiazole hybrids through phenyl linked thiosemicarbazide antileishmanial scaffolds: In silico validation and SAR studies. Bioorg. Chem. 2017, 71, 192–200. [Google Scholar] [CrossRef]
- Opo, F.A.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Saeed, F.; Salim, N.; Abdo, A.; Hentabli, H. Graph-based consensus clustering for combining multiple clusterings of chemical structures. Mol. Inform. 2013, 32, 165–178. [Google Scholar] [CrossRef]
- Elkady, H.; Elwan, A.; El-Mahdy, H.A.; Doghish, A.S.; Ismail, A.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Dahab, M.A.; Mahdy, H.A. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzyme Inhib. Med. Chem. 2022, 37, 397–410. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Elkady, H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Al-Hossaini, A.M.; Alharbi, M.A.; Eissa, I.H.; Dahab, M.A. Discovery of new quinoxaline-based derivatives as anticancer agents and potent VEGFR-2 inhibitors: Design, synthesis, and in silico study. J. Mol. Struct. 2022, 1253, 132220. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Elkady, H.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Alanazi, M.M.; Alharbi, M.A.; Eissa, I.H.; Dahab, M.A. New quinoxaline-based VEGFR-2 inhibitors: Design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies. RSC Adv. 2021, 11, 30315–30328. [Google Scholar] [CrossRef]
- El-Helby, A.G.A.; Ayyad, R.R.; El-Adl, K.; Elkady, H. Phthalazine-1, 4-dione derivatives as non-competitive AMPA receptor antagonists: Design, synthesis, anticonvulsant evaluation, ADMET profile and molecular docking. Mol. Divers. 2019, 23, 283–298. [Google Scholar] [CrossRef]
- El-Helby, A.G.A.; Ayyad, R.R.; Zayed, M.F.; Abulkhair, H.S.; Elkady, H.; El-Adl, K. Design, synthesis, in silico ADMET profile and GABA-A docking of novel phthalazines as potent anticonvulsants. Arch. Der Pharm. 2019, 352, 1800387. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Ishmuratova, M.Y.; Toppet, S.; Dehaen, W.; Alsfouk, A.A.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M. Isolation and In Silico SARS-CoV-2 Main Protease Inhibition Potential of Jusan Coumarin, a New Dicoumarin from Artemisia glauca. Molecules 2022, 27, 2281. [Google Scholar] [CrossRef]
- Jalmakhanbetova, R.I.; Suleimen, Y.M.; Oyama, M.; Elkaeed, E.B.; Eissa, I.; Suleimen, R.N.; Metwaly, A.M.; Ishmuratova, M.Y. Isolation and in silico anti-COVID-19 main protease (Mpro) activities of flavonoids and a sesquiterpene lactone from Artemisia sublessingiana. J. Chem. 2021, 2021, 5547013. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biovia. Qsar, Admet and Predictive Toxicology. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 15 February 2022).
- Elkaeed, E.B.; Elkady, H.; Belal, A.; Alsfouk, B.A.; Ibrahim, T.H.; Abdelmoaty, M.; Arafa, R.K.; Metwaly, A.M.; Eissa, I.H. Multi-Phase In Silico Discovery of Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors among 3009 Clinical and FDA-Approved Related Drugs. Processes 2022, 10, 530. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Li, C.-X.; Wang, H.-B.; Oppong, D.; Wang, J.-X.; Chen, J.-F.; Le, Y. Excipient-assisted vinpocetine nanoparticles: Experiments and molecular dynamic simulations. Mol. Pharm. 2014, 11, 4023–4035. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. Open Source Drug Discovery Consortium. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Hagras, M.; El Deeb, M.A.; Elzahabi, H.S.; Elkaeed, E.B.; Mehany, A.B.; Eissa, I.H. Discovery of new quinolines as potent colchicine binding site inhibitors: Design, synthesis, docking studies, and anti-proliferative evaluation. J. Enzyme Inhib. Med. Chem. 2021, 36, 640–658. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; Abdelhady, A.A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Mahdy, H.A. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorg. Chem. 2021, 107, 104532. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Mahdy, H.A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Dahab, M.A.; Eissa, I.H. New bis ([1, 2, 4] triazolo) [4, 3-a: 3′, 4′-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation. Bioorg. Chem. 2021, 112, 104949. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Alesawy, M.S.; Eissa, S.I.; El-Fakharany, E.M.; Kalaba, M.H.; Sharaf, M.H.; Shama, N.M.A.; Mahmoud, S.H.; Mostafa, A.; Al-Karmalawy, A.A. Design and synthesis of new 4-(2-nitrophenoxy) benzamide derivatives as potential antiviral agents: Molecular modeling and in vitro antiviral screening. New J. Chem. 2021, 45, 16557–16571. [Google Scholar] [CrossRef]
- Yousef, R.; Sakr, H.; Eissa, I.; Mehany, A.; Metwaly, A.; Elhendawy, M.A.; Radwan, M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Amer, H.H.; Alotaibi, S.H.; Trawneh, A.H.; Metwaly, A.M.; Eissa, I.H. Anticancer activity, spectroscopic and molecular docking of some new synthesized sugar hydrazones, Arylidene and α-Aminophosphonate derivatives. Arab. J. Chem. 2021, 14, 103348. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Al-Karmalawy, A.A.; Elkaeed, E.B.; Alswah, M.; Belal, A.; Taghour, M.S.; Eissa, I.H. Design and discovery of new 1, 2, 4-triazolo [4, 3-c] quinazolines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Der Pharm. 2021, 354, 2000237. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | ALog p | M. Wt | HBA | HBD | Rotatable Bonds | Rings | Aromatic Rings | MFPSA | Minimum Distance |
|---|---|---|---|---|---|---|---|---|---|
| 3 | 2.91 | 313.348 | 4 | 3 | 6 | 2 | 2 | 0.239 | 1.802 |
| 7 | 0.857 | 546.629 | 9 | 3 | 11 | 5 | 1 | 0.223 | 1.053 |
| 17 | 3.911 | 444.522 | 4 | 2 | 11 | 3 | 3 | 0.183 | 1.382 |
| 49 | 3.714 | 481.647 | 5 | 1 | 7 | 5 | 2 | 0.11 | 1.682 |
| 50 | 3.387 | 465.604 | 5 | 2 | 6 | 5 | 2 | 0.132 | 1.689 |
| 85 | 0.436 | 446.404 | 10 | 5 | 5 | 4 | 2 | 0.373 | 1.801 |
| 94 | 3.743 | 372.412 | 6 | 1 | 7 | 3 | 2 | 0.192 | 1.804 |
| 95 | 2.879 | 388.411 | 7 | 2 | 7 | 3 | 2 | 0.235 | 1.671 |
| 96 | 3.513 | 444.431 | 9 | 0 | 10 | 4 | 2 | 0.225 | 1.515 |
| 97 | 3.474 | 442.415 | 9 | 0 | 9 | 4 | 2 | 0.226 | 1.586 |
| 128 | −0.546 | 798.738 | 20 | 10 | 16 | 5 | 2 | 0.407 | 0.703 |
| 129 | −0.925 | 756.702 | 19 | 11 | 14 | 5 | 2 | 0.425 | 0.795 |
| 130 | −0.699 | 770.728 | 19 | 10 | 15 | 5 | 2 | 0.396 | 0.704 |
| 131 | 0.484 | 624.587 | 15 | 9 | 11 | 4 | 2 | 0.414 | 0.726 |
| 132 | 0.484 | 624.587 | 15 | 9 | 11 | 4 | 2 | 0.413 | 0.725 |
| 156 | 2.489 | 464.635 | 6 | 5 | 5 | 4 | 0 | 0.236 | 0.888 |
| 157 | 1.195 | 480.634 | 7 | 6 | 5 | 4 | 0 | 0.27 | 0.884 |
| 158 | 1.137 | 480.634 | 7 | 6 | 5 | 4 | 0 | 0.266 | 0.886 |
| 180 | 3.176 | 944.185 | 12 | 5 | 13 | 3 | 1 | 0.232 | 0.674 |
| 184 | 4.986 | 836.071 | 8 | 6 | 11 | 4 | 2 | 0.23 | 0.623 |
| 203 | −0.499 | 503.583 | 10 | 4 | 8 | 3 | 0 | 0.268 | 1.002 |
| 204 | −0.091 | 517.61 | 10 | 3 | 9 | 3 | 0 | 0.237 | 0.999 |
| 210 | 3.874 | 512.638 | 6 | 1 | 13 | 3 | 2 | 0.188 | 0.902 |
| 237 | 3.511 | 557.073 | 9 | 2 | 8 | 4 | 0 | 0.235 | 1.025 |
| 248 | 3.607 | 605.642 | 7 | 3 | 7 | 4 | 0 | 0.156 | 1.038 |
| 264 | 5.732 | 470.598 | 6 | 2 | 10 | 2 | 1 | 0.172 | 0.723 |
| 276 | 3.062 | 557.03 | 10 | 1 | 19 | 1 | 0 | 0.24 | 0.717 |
| 277 | 1.804 | 754.797 | 14 | 5 | 6 | 4 | 2 | 0.295 | 0.604 |
| 278 | 2.745 | 811.868 | 15 | 6 | 6 | 4 | 1 | 0.288 | 0.692 |
| 280 | −0.367 | 480.523 | 5 | 7 | 10 | 3 | 3 | 0.359 | 0.649 |
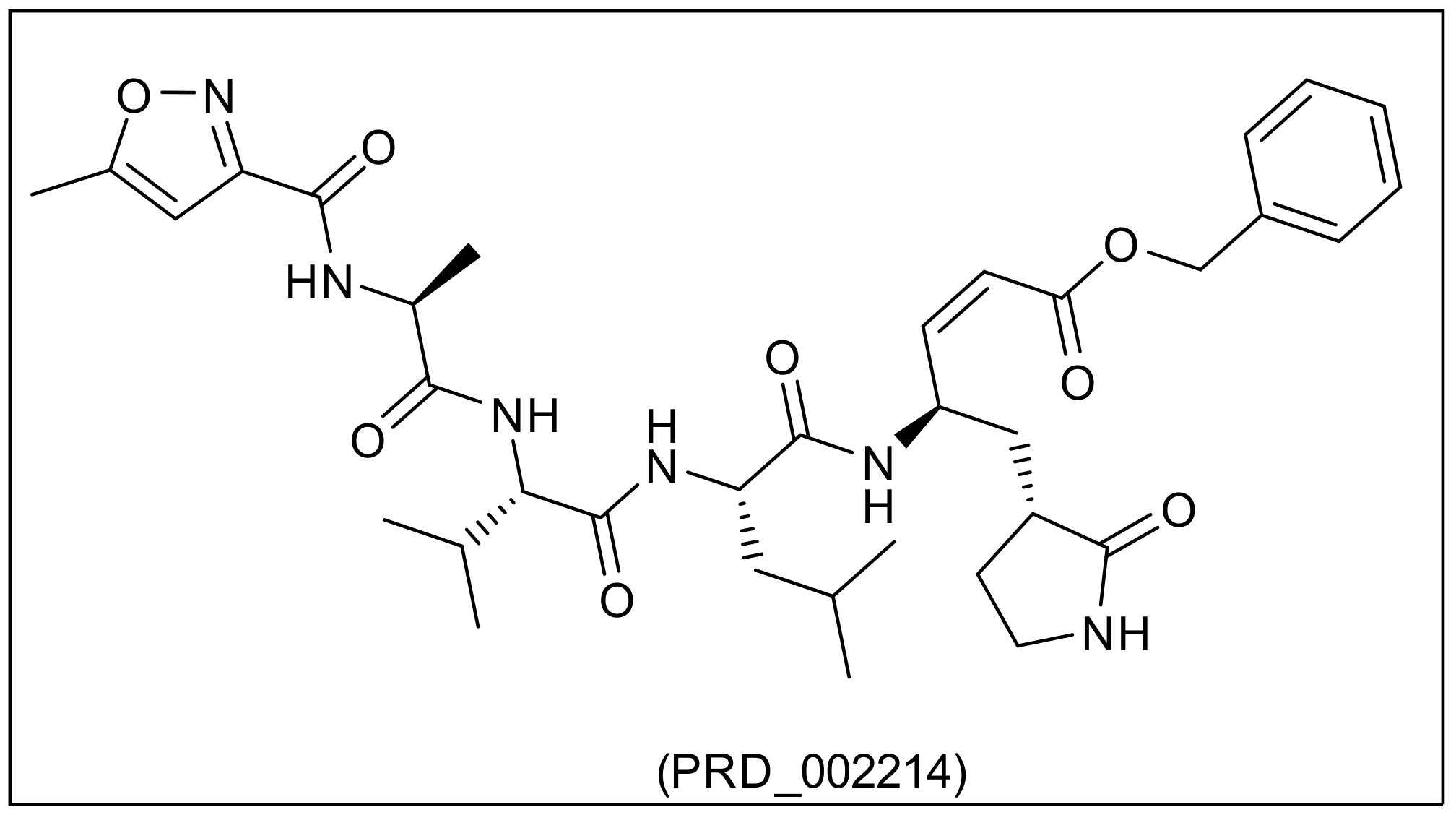
| PRD_002214 | 2.453 | 680.791 | 8 | 5 | 18 | 3 | 2 | 0.273 | - |
| No. | Name and Type | Source | Antiviral Activity |
|---|---|---|---|
| 3 | Moupinamide, an alkaloid | Mollinedia sp. [59] | Showed in silico inhibition against Mpro 6Y84 and the spike protein 6LXT) [60] |
| 7 | Homoharringtonine, an alkaloid | Cephalotaxus genus [61] | Inhibited the replication of SARS-CoV-2 (in vitro) with an EC50 value of 2.55 μM [62] |
| 17 | Aurantiamide acetate, a dipeptide | Pongamia glabra flowers [63] and Aspergillus sp [64] | In vitro inhibited the replication of Influenza A virus in MDCK cells [65] |
| 49 | Emetine, an alkaloid | Cephaelis ipecacuanha roots [66] | Inhibited SARS-CoV-2 replication in vitro with an EC50 of 0.46 μM [62] Inhibited SARS-CoV-2 protein synthesis and interaction of viral mRNA [67] |
| 50 | Psychotrine, an alkaloid | Cephaelis acuminata [68] | Inhibited COVID-19 Mpro in silico (ΔG = −3.5 kcal. mol−1) [69] |
| 85 | 5-O-Methylgenistein-7-glucoside, an isoflavonoid | Ulex europaeus [70] | Inhibited herpes simplex virus (HSV) in vitro [70] |
| 94 | Arctigenin, a lignan | Arctium lappa [71] | In vivo inhibited influenza virus through interferon production [72]. Inhibited Spring viraemia of carp virus (SVCV) through inhibition of autophagy [73] |
| 95 | Trachelogenin, a lignan | Ipomoea cairica [74] | Inhibited the entry of hepatitis C virus through CD81 [75] |
| 96 | Rhinacanthin-F, a lignan | Rhinacanthus nasutus [76] | Inhibited of influenza virus type A [76] |
| 97 | Rhinacanthin-E, a lignan | ||
| 128 | Luteosides A, B and C phenylpropanoid glycosides | Markhamia lutea [77] | Showed an in vitro inhibition of respiratory syncytial virus [77] |
| 129 | |||
| 130 | |||
| 131 | Verbascoside, a phenylpropanoid | Verbascum olympicum [78] and Markhamia lutea [77] | Inhibited in vitro herpes HSV-1, HSV-2 [79] and a respiratory syncytial virus [77] |
| 132 | Isoverbascoside, a phenylpropanoid | In vitro inhibited the respiratory syncytial virus [77] | |
| 156 | Ponasterone A, a triterpenoidal saponins | Podocarpus macrophyllus [80] Acrostichum aureum [81] | Inhibited HIV-1 gene expression in mammalian cells [82] |
| 157 | Pterosterone, a triterpenoidal saponins | Exhibited an inhibition against (HIV-1) infection as CCR5 inhibitors [83] | |
| 158 | Ecdysterone, a riterpenoidal saponins | Diploclisia glaucescens [84] | Inhibited HIV-1 in vitro [70] |
| 180 | Didemnin A, a peptide (depsipeptide) | Caribbean tunicate Trididemnum solidum [85] | Inhibited Coxsackie virus and equine rhinovirus in vitro [85] Inhibited both RNA and DNA viruses and HSV-2 in vitro [86] |
| 184 | Kahalalide E, a peptide | Marine Mollusk Elysia rufescens [87] | Inhibited HSV-2 in vitro [88] |
| 203 | Mycalamide A, an alkaloid | of the genus Mycale [89] | Inhibited SARS-CoV-1 in vitro with an IC50 of 0.2 µg kg−1 [90] and at a concentration of 5 ng/disc it stopped HSV-1 and Polio type I viruses [91] |
| 204 | Mycalamide B, an alkaloid | At a concentration of 2 ng/disc, it stopped HSV-1 and Polio-1 viruses [91] | |
| 210 | Hennoxazole A, an alkaloid | A sponge Polyfibrospongia sp [92] | In vitro inhibited HSV-1 (IC50 of 0.6 lg/mL) [92] |
| 237 | Solenolide A, a diterpene | Marine Octocoral of the Genus Solenopodium [93] | In silico inhibition of Mpro PDB Id: 6LU7 with a binding free energy of −10.8 kcal. mol−1 [94] |
| 248 | Thyrsiferol, a triterpene | The red algae Laurencia thyrsifera [95] | In vitro inhibited VSV and HSV-1 at levels of 0.l-0μg/well and slight activity against A59 coronavirus [96] |
| 264 | Usneoidol Z a meroterpene | Brown Seaweed Cystoseira usneoides [97,98] | In vitro inhibited HSV-l/CV-I at concentrations of 20 and 10 μg/disk, respectively [98] |
| 276 | Punaglandin-1, an eicosanoid | The octocoral Telesto riisei [99] | Inhibited HSV in vitro [70] |
| 277 | Rifamycin B, a macrolide | The bacterium Amycolatopsis rifamycinica [100] | Inhibited (in vitro) murine sarcoma virus through cell transformation inhibition [101] |
| 278 | Streptovaricin B, an ansamycin | Streptomyces spectabilis, an actinomycete [102] | Stopped poxviruses replication through the inhibition of mRNA synthesis in early stages [101] |
| 280 | Distamycin A, an oligopeptide | Streptomyces netropsis [103] | Inhibited transcription and replication of different viruses [104] and inhibited the post-replicative mRNA synthesis of vaccinia virus [105] |
| Comp. | Similarity | SA | SB | SC |
|---|---|---|---|---|
| PRD_002214 | 1 | 1116 | 0 | 0 |
| 7 | 0.683 | 772 | 15 | 344 |
| 128 | 0.648 | 926 | 313 | 190 |
| 130 | 0.651 | 889 | 249 | 227 |
| 156 | 0.652 | 818 | 139 | 298 |
| 157 | 0.654 | 824 | 143 | 292 |
| 158 | 0.644 | 819 | 156 | 297 |
| 180 | 0.718 | 1509 | 987 | −393 |
| 184 | 0.800 | 1372 | 599 | −256 |
| 203 | 0.654 | 755 | 39 | 361 |
| 204 | 0.644 | 780 | 95 | 336 |
| 210 | 0.666 | 748 | 7 | 368 |
| 237 | 0.676 | 868 | 168 | 248 |
| 264 | 0.681 | 725 | −52 | 391 |
| 276 | 0.665 | 859 | 176 | 257 |
| 277 | 0.758 | 1026 | 237 | 90 |
| 278 | 0.724 | 1207 | 550 | −91 |
| Compound | ∆G (kcal. mol−1) | Compound | ∆G (kcal. mol−1) |
|---|---|---|---|
| 7 | −25.20 | 204 | −33.03 |
| 128 | −29.53 | 210 | −28.41 |
| 130 | −32.99 | 237 | −23.75 |
| 156 | −29.09 | 264 | −25.85 |
| 157 | −24.19 | 276 | −21.66 |
| 158 | −26.98 | 277 | −24.08 |
| 180 | −34.15 | 278 | −29.00 |
| 184 | −30.15 | PRD_002214 | −31.31 |
| 203 | −31.20 |
| Comp. | 128 | 130 | 156 | 180 | 184 | 203 | 204 | 278 | Simeprevir |
|---|---|---|---|---|---|---|---|---|---|
| FDA rodent carcinogenicity | Non-Carcinogen | ||||||||
| Median carcinogenic potency (TD50), mg/kg/day | 2.871 | 1.854 | 5.663 | 8.687 | 3.037 | 7.360 | 12.564 | 12.946 | 2.014 |
| Rat maximum tolerated dose, g/kg body weight | 2.382 | 1.277 | 0.137 | 0.002 | 0.021 | 0.018 | 0.029 | 0.020 | 0.003 |
| Rat lethal dose (LD50) g/kg body weight | 4.282 | 5.717 | 10.020 | 0.274 | 4.897 | 0.141 | 0.324 | 0.166 | 0.209 |
| Rat chronic lowest observed adverse effect level (LOAEL), g/kg body weight | 0.040 | 0.017 | 0.017 | 0.001 | 0.012 | 0.001 | 0.001 | 0.001 | 0.002 |
| Ocular irritancy | Mild | Mild | Moderate | Moderate | None | Mild | Mild | Mild | Mild |
| Skin irritancy | Mild | Mild | Moderate | Mild | None | Mild | Mild | None | None |
| Complex | ΔE Binding (kj/mol) | ΔE Electrostatic (kj/mol) | ΔE Vander Waal (kj/mol) | ΔE Polar Solvation (kj/mol) | SASA (kJ/mol) |
|---|---|---|---|---|---|
| 130 | −286.9 ± 10.2 | −139.1 ± 9.8 | −245.7 ± 12.3 | 125.2 ± 8.1 | −27.3 ± 0.9 |
| 184 | −271.8 ± 8.9 | −131.1 ± 9.1 | −238.9 ± 8.5 | 124.9 ± 6.2 | −26.7 ± 1.1 |
| 278 | −236.6 ± 10.4 | −111.8 ± 11.3 | −205.6 ± 9.4 | 102.1 ± 10.7 | −21.3 ± 0.8 |
| PRD_002214 | −252.5 ± 9.1 | −119.5 ± 8.7 | −226.7 ± 10.9 | 114.2 ± 7.4 | −20.5 ± 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkaeed, E.B.; Youssef, F.S.; Eissa, I.H.; Elkady, H.; Alsfouk, A.A.; Ashour, M.L.; El Hassab, M.A.; Abou-Seri, S.M.; Metwaly, A.M. Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease. Int. J. Mol. Sci. 2022, 23, 6912. https://doi.org/10.3390/ijms23136912
Elkaeed EB, Youssef FS, Eissa IH, Elkady H, Alsfouk AA, Ashour ML, El Hassab MA, Abou-Seri SM, Metwaly AM. Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease. International Journal of Molecular Sciences. 2022; 23(13):6912. https://doi.org/10.3390/ijms23136912
Chicago/Turabian StyleElkaeed, Eslam B., Fadia S. Youssef, Ibrahim H. Eissa, Hazem Elkady, Aisha A. Alsfouk, Mohamed L. Ashour, Mahmoud A. El Hassab, Sahar M. Abou-Seri, and Ahmed M. Metwaly. 2022. "Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease" International Journal of Molecular Sciences 23, no. 13: 6912. https://doi.org/10.3390/ijms23136912